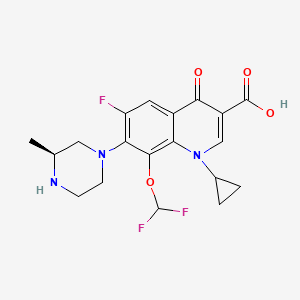
![str1]()
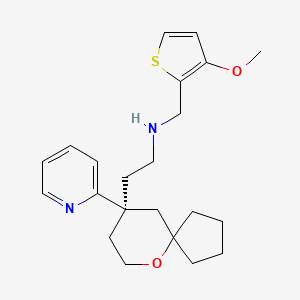
VX-?
An Azaindolyl-Pyrimidine Inhibitor of Influenza Virus Replication from Vertex
SYNTHESIS COMING……..
CAS 1259498-06-0
Specific Rotation
[α]21D = −165.7° (c = 1 in MeOH).
1H NMR (300 MHz, d6-DMSO) δ 12.23 (s, 1H), 8.42 (dd, J = 9.8, 2.9 Hz, 1H), 8.34–8.18 (m, 2H), 8.14 (d, J = 4.0 Hz, 1H), 7.49 (d, J = 7.5 Hz, 1H), 6.33 (d, J= 7.6 Hz, 1H), 4.24–4.00 (m, 1H), 3.75–3.57 (m, 1H), 3.57–3.42 (m, 4H), 3.28–3.09 (m, 4H), 2.15 (d, J = 11.4 Hz, 1H), 2.01 (d, J = 11.2 Hz, 1H), 1.83 (d, J = 9.7 Hz, 2H), 1.60–1.07 (m, 4H).19F NMR (282.4 MHz, d6-DMSO) −138.10, −158.25 ppm.
HRMS (ESI) [M + H]+ calculated for C22H26F2N7O2 458.2111, found 458.2110.
Influenza spreads around the world in seasonal epidemics, resulting in the deaths of hundreds of thousands annually – millions in pandemic years. For example, three influenza pandemics occurred in the 20th century and killed tens of millions of people, with each of these pandemics being caused by the appearance of a new strain of the virus in humans. Often, these new strains result from the spread of an existing influenza virus to humans from other animal species.
Influenza is primarily transmitted from person to person via large virus-laden droplets that are generated when infected persons cough or sneeze; these large droplets can then settle on the mucosal surfaces of the upper respiratory tracts of susceptible individuals who are near (e.g. within about 6 feet) infected persons. Transmission might also occur through direct contact or indirect contact with respiratory secretions, such as touching surfaces contaminated with influenza virus and then touching the eyes, nose or mouth. Adults might be able to spread influenza to others from 1 day before getting symptoms to approximately 5 days after symptoms start. Young children and persons with weakened immune systems might be infectious for 10 or more days after onset of symptoms. [00103] Influenza viruses are RNA viruses of the family Orthomyxoviridae, which comprises five genera: Influenza virus A, Influenza virus B, Influenza virus C, Isavirus and Thogoto virus.
The Influenza virus A genus has one species, influenza A virus. Wild aquatic birds are the natural hosts for a large variety of influenza A. Occasionally, viruses are transmitted to other species and may then cause devastating outbreaks in domestic poultry or give rise to human influenza pandemics. The type A viruses are the most virulent human pathogens among the three influenza types and cause the most severe disease. The influenza A virus can be subdivided into different serotypes based on the antibody response to these viruses. The serotypes that have been confirmed in humans, ordered by the number of known human pandemic deaths, are: HlNl (which caused Spanish influenza in 1918), H2N2 (which caused Asian Influenza in 1957), H3N2 (which caused Hong Kong Flu in 1968), H5N1 (a pandemic threat in the 2007-08 influenza season), H7N7 (which has unusual zoonotic potential), H1N2 (endemic in humans and pigs), H9N2, H7N2 , H7N3 and H10N7. [00105] The Influenza virus B genus has one species, influenza B virus. Influenza B almost exclusively infects humans and is less common than influenza A. The only other animal known to be susceptible to influenza B infection is the seal. This type of influenza mutates at a rate 2-3 times slower than type A and consequently is less genetically diverse, with only one influenza B serotype. As a result of this lack of antigenic diversity, a degree of immunity to influenza B is usually acquired at an early age. However, influenza B mutates enough that lasting immunity is not possible. This reduced rate of antigenic change, combined with its limited host range (inhibiting cross species antigenic shift), ensures that pandemics of influenza B do not occur.
The Influenza virus C genus has one species, influenza C virus, which infects humans and pigs and can cause severe illness and local epidemics. However, influenza C is less common than the other types and usually seems to cause mild disease in children. [00107] Influenza A, B and C viruses are very similar in structure. The virus particle is 80-120 nanometers in diameter and usually roughly spherical, although filamentous forms can occur. Unusually for a virus, its genome is not a single piece of nucleic acid; instead, it contains seven or eight pieces of segmented negative-sense RNA. The Influenza A genome encodes 11 proteins: hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), Ml, M2, NSl, NS2(NEP), PA, PBl, PB1-F2 and PB2.
HA and NA are large glycoproteins on the outside of the viral particles. HA is a lectin that mediates binding of the virus to target cells and entry of the viral genome into the target cell, while NA is involved in the release of progeny virus from infected cells, by cleaving sugars that bind the mature viral particles. Thus, these proteins have been targets for antiviral drugs. Furthermore, they are antigens to which antibodies can be raised. Influenza A viruses are classified into subtypes based on antibody responses to HA and NA, forming the basis of the H and N distinctions (vide supra) in, for example, H5N1. [00109] Influenza produces direct costs due to lost productivity and associated medical treatment, as well as indirect costs of preventative measures. In the United States, influenza is responsible for a total cost of over $10 billion per year, while it has been estimated that a future pandemic could cause hundreds of billions of dollars in direct and indirect costs. Preventative costs are also high. Governments worldwide have spent billions of U.S. dollars preparing and planning for a potential H5N1 avian influenza pandemic, with costs associated with purchasing drugs and vaccines as well as developing disaster drills and strategies for improved border controls.
Current treatment options for influenza include vaccination, and chemotherapy or chemoprophylaxis with anti-viral medications. Vaccination against influenza with an influenza vaccine is often recommended for high-risk groups, such as children and the elderly, or in people that have asthma, diabetes, or heart disease. However, it is possible to get vaccinated and still get influenza. The vaccine is reformulated each season for a few specific influenza strains but cannot possibly include all the strains actively infecting people in the world for that season. It takes about six months for the manufacturers to formulate and produce the millions of doses required to deal with the seasonal epidemics; occasionally, a new or overlooked strain becomes prominent during that time and infects people although they have been vaccinated (as by the H3N2 Fujian flu in the 2003-2004 influenza season). It is also possible to get infected just before vaccination and get sick with the very strain that the vaccine is supposed to prevent, as the vaccine takes about two weeks to become effective. [00111] Further, the effectiveness of these influenza vaccines is variable. Due to the high mutation rate of the virus, a particular influenza vaccine usually confers protection for no more than a few years. A vaccine formulated for one year may be ineffective in the following year, since the influenza virus changes rapidly over time, and different strains become dominant.
Also, because of the absence of RNA proofreading enzymes, the RNA- dependent RNA polymerase of influenza vRNA makes a single nucleotide insertion error roughly every 10 thousand nucleotides, which is the approximate length of the influenza vRNA. Hence, nearly every newly-manufactured influenza virus is a mutant — antigenic drift. The separation of the genome into eight separate segments of vRNA allows mixing or reassortment of vRNAs if more than one viral line has infected a single cell. The resulting rapid change in viral genetics produces antigenic shifts and allows the virus to infect new host species and quickly overcome protective immunity.
Antiviral drugs can also be used to treat influenza, with neuraminidase inhibitors being particularly effective, but viruses can develop resistance to the standard antiviral drugs.
PAPER
http://pubs.acs.org/doi/full/10.1021/acs.oprd.6b00063
Development of a Scalable Synthesis of an Azaindolyl-Pyrimidine Inhibitor of Influenza Virus Replication
Vertex Pharmaceuticals Incorporated, 50 Northern Avenue, Boston, Massachusetts 02210, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00063
Publication Date (Web): April 08, 2016
A scalable, asymmetric route for the synthesis of the influenza virus replication inhibitor 2 is presented. The key steps include an enzymatic desymmetrization of cis-1,3-cyclohexanediester in 99% yield and 96% ee, SNAr displacement of a methanesulfinylpyrimidine, and a Curtius rearrangement to form a morpholinyl urea. This high-yielding route allowed us to rapidly synthesize hundreds of grams of 2 in 99% purity to support in vivo studies.
![]()
About Influenza
Often called “the flu,” seasonal influenza is caused by influenza viruses, which infect the respiratory tract.1 The flu can result in seasonal epidemics2 and can produce severe disease and high mortality in certain populations, such as the elderly.3 Each year, on average 5 to 20 percent of the U.S. population gets the flu4 resulting in more than 200,000 flu-related hospitalizations and 36,000 deaths.5 The overall national economic burden of influenza-attributable illness for adults is $83.3 billion.5 Direct medical costs for influenza in adults totaled $8.7 billion including $4.5 billion for adult hospitalizations resulting from influenza-attributable illness.5 The treatment of the flu consists of antiviral medications that have been shown in clinical studies to shorten the disease and reduce the severity of symptoms if taken within two days of infection.6 There is a significant need for new medicines targeting flu that provide a wider treatment window, greater efficacy and faster onset of action.
About Vertex
Vertex is a global biotechnology company that aims to discover, develop and commercialize innovative medicines so people with serious diseases can lead better lives. In addition to our clinical development programs focused on cystic fibrosis, Vertex has more than a dozen ongoing research programs aimed at other serious and life-threatening diseases.
Founded in 1989 in Cambridge, Mass., Vertex today has research and development sites and commercial offices in the United States, Europe, Canada and Australia. For four years in a row, Science magazine has named Vertex one of its Top Employers in the life sciences. For additional information and the latest updates from the company, please visit www.vrtx.com.
Vertex’s press releases are available at www.vrtx.com.
![]()
![]()
![str1]()
SYNTHESIS COMING
WO-2010148197
http://www.google.co.in/patents/WO2010148197A1?cl=en
General Scheme 44 SIMILAR TO A POINT BUT NOT SAME
(a) Pd(PPh3)4 sodium carbonate, DME/water, reflux (b) meta-chloroperbenzoic acid, dichloromethane, rt. (c) 20a, tetrahydrofuran, 5O°C (d) trifluoroacetic acid, dichloromethane, rt.
SIMILAR NOT SAME
(e) morpholιne-4-carbonyl chloride, dimethylformamide, rt (f) sodium methoxide, methanol, rt.
Formation of 5-fluoro-3-[5-fluoro-4-(methylthio)pyrimidin-2-yl]-1-tosyl-lΗ- pyrrolo[2,3-b]pyridine (44b)
2-Chloro-5-fluoro-4-methylsulfanyl-pyrimidine (34.1 g, 191.0 mmol) , 5-fluoro-1-(p- tolylsulfonyl)-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyrrolo[2,3-b]pyridine, 44a, (53.0 g, 127.3 mmol) and Na2Cθ3 (40.5 g, 381.9 mmol) were dissolved in a mixture of DME (795 mL) and water (159 mL). The mixture was purged with nitrogen for 20 minutes and treated with Pd(PPh3 )4 (7.4 g, 6.6 mmol). After purging with nitrogen for another 20 minutes, the reaction was heated to reflux overnight, cooled to room temperature and diluted with water (60OmL). The resulting suspension was stirred at room temperature for 30 minutes and the precipitate was then collected by filtration, washed with water and acetonitrile and dried at 50 °C to afford 48.2 g of 5-fluoro-3-[5-fluoro-4-(methylthio)pyrimidin-2-yl]-1-tosyl-1H- pyrrolo[2,3-b]pyridine as a white solid.
1H NMR (300 MHz, OMSO-d6) δ 8.70 – 8.58 (m, 2H), 8.54 – 8.41 (m, 2H), 8.09 (d, J = 8.4 Hz, 2H), 7.45 (d, J= 8.2 Hz, 2H), 2.76 (s, 3H), 2.36 (s, 3H).
Formation of 5-fluoro-3-[5-fluoro-4-(methylsulfinyl)pyrimidin-2-yl]-1- tosyl-1H-pyrrolo[2,3-b]pyridine (44c)
5-fluoro-3 – [5 -fluoro-4-(methylthio)pyrimidin-2-yl] – 1 -tosyl- 1 H-pyrrolo [2,3 – b]pyridine, 44b, (48.2 g, 111.5 mmol) was dissolved in dichloromethane (2.3 L) and treated portionwise with m-CPBA (27.5 g, 122.6 mmol) while keeping the temperature below 20 °C. After addition was complete, the reaction was stirred at room temperature for 2 hours, then treated with another portion of m-CPBA (1.9 g) and stirred for another hour. The reaction mixture was washed with 12% aqueuous K2CO3 (2 x 1.0 L) and the organic layer was dried on Na2SO4 and concentrated in vacuo to provide 50 g of 5-fluoro-3-[5-fluoro-4- (methylsulfinyl)pyrimidin-2-yl]-1-tosyl-1H-pyrrolo[2,3-b]pyridine as a yellow solid.
1H NMR (300 MHz, DMSO-rf<5) δ 9.11 (d, J= 1.5 Hz, 1H), 8.69 (s, 1H), 8.65 (dd, J = 9.0, 2.9 Hz, 1H), 8.52 (dd, J= 2.8, 1.2 Hz, 1H), 8.11 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.3 Hz, 2H), 3.05 (s, 3H), 2.36 (s, 3H).
[001057] Formation of tert-butyl N-[(IR, 3S)-3-[[5-fluoro-2-[5-fluoro-1-(p- tolylsulfonyl)pyrrolo [2,3-b] pyridin-3-yl]pyrimidin-4-yl] amino] cyclohexyl] carbamate (44d)
5-fluoro-3-(5-fluoro-4-methylsulfinyl-pyrimidin-2-yl)-1-(p-tolylsulfonyl)pyrrolo[2,3- b]pyridine, 44c, (5.9 g, 10.5 mmol) and tert-butyl N-[(IR, 35*)-3-aminocyclohexyl]carbamate (3 g, 12.60 mmol) were dissolved in THF (100 mL). The reaction mixture was heated to 50 °C for 6 hours, then cooled to room temperature. C6 lite was added and the solvent was removed under reduced pressure. The C6 lite-supported residue was purified by silica gel chromatography (20-80% EtOAc/hexanes gradient to provide 3.7 g of tert-butyl N-[(IR, 3S)- 3-[[5-fluoro-2-[5-fluoro-1-(p-tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4- yl]amino]cyclohexyl]carbamate.
1H NMR (300 MHz, CDCl3) δ 8.51 (s, 1H), 8.46 – 8.41 (m, 1H), 8.29 (d, J = 1.6 Hz, 1H), 8.11 (s, 1H), 8.08 (s, 1H), 8.06 (d, J= 3.2 Hz, 1H), 7.27 (d, J= 8.4 Hz, 2H), 4.91 (d, J = 8.0 Hz, 1H), 4.41 (s, 1H), 4.29 – 4.01 (m, 1H), 3.64 (s, 1H), 2.47 (d, J= 11.5 Hz, 1H), 2.36 (s, 3H), 2.24 (d, J = 13.1 Hz, 1H), 2.08 (d, J= 10.9 Hz, 1H), 1.91 (d, J= 13.8 Hz, 1H), 1.43 (s, 9H), 1.30 – 1.03 (m, 4H).
Formation of (IS, SΛHVHS-fluoro^-β-fluoro-1-Cp- tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4-yl]cyclohexane-1,3-diamine (44e) tert-Butyl N-[(IR, 3S>3-[[5-fluoro-2-[5-fluoro-1-(p-tolylsulfonyl)pyrrolo[2,3- b]pyridin-3-yl]pyrimidin-4-yl]amino]cyclohexyl]carbamate, 44d, (3.7 g, 6.2 mmol) was dissolved in dichloromethane (105 mL) and treated with trifluoroacetic acid (31 mL). After 5 minutes, the volatiles were evaporated under reduced pressure, and the resulting residue was treated with IN NaOH (75 mL). The resulting precipitate was collected by filtration, washed with water (3 x 30 mL) and vacuum dried to provide 2.7 g of (IS, 3R)-Nl -[5-fluoro-2-[5- fluoro-1-(p-tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4-yl]cyclohexane-l,3-diamine as a white solid.
1H NMR (300 MHz, MeOD) d 8.56 (dd, J = 8.0, 3.9 Hz, 2H), 8.35 – 8.26 (m, 1H), 8.12 (dd, J= 10.3, 6.1 Hz, 3H), 7.43 (d, J= 8.4 Hz, 2H), 4.36 – 4.21 (m, 1H), 3.28 – 3.13 (m, 1H), 2.48 (d, J= 12.3 Hz, 1H), 2.46 (s, 3H), 2.25 – 1.97 (m, J= 17.3, 10.6, 4.1 Hz, 4H), 1.76 – 1.28 (m, 3H).
Formation of N-[(IR, 3S>3-[[5-fluoro-2-[5-fluoro-1-(p- tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4-yl]amino]cyclohexyl] morpholine- 4-carboxamide (44f)
(15, 3R)-M-[5-fluoro-2-[5-fluoro-1-(p-tolylsulfonyl)pyrrolo[2,3-b]pyridin-3- yl]pyrimidin-4-yl]cyclohexane- 1,3 -diamine, 44e, (2.3 g, 4.6 mmol) was dissolved in DMF (5OmL) and treated with morpholine-4-carbonyl chloride (2.1 g, 13.8 mmol) and DIPEA (4.2 g, 5.6 mL, 32.3 mmol). After one hour, the resulting solution was diluted with water (400 mL) and stirred for an additional two hours. The resulting precipitate was collected by filtration, washed with water (3 x 50 mL) and dried to provide the crude product. This material was purified by flash chromatography on a 4Og column using EtOAc/DCM 20- 100%, to provide 2.0 g of N-[(1R, 35)-3-[[5-fluoro-2-[5-fluoro-1-(p- tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4-yl]amino]cyclohexyl]morpholine-4- carboxamide as a white solid.
1H NMR (300 MHz, DMSO-Λ5) δ 8.53 – 8.43 (m, J = 11.9, 2.7 Hz, 3H), 8.22 (d, J = 3.9 Hz, 1H), 8.07 (d, J= 8.4 Hz, 2H), 7.44 (d, J= 8.3 Hz, 2H), 6.32 (d, J= 7.5 Hz, 1H), 4.05 (s, J= 19.4 Hz, 1H), 3.62 (s, 1H), 3.58 – 3.45 (m, 4H), 3.27 – 3.18 (m, 4H), 2.36 (s, 3H), 2.12 (d, J= 11.7 Hz, 1H), 1.99 (d, J= 9.5 Hz, 1H), 1.83 (d, J= 10.3 Hz, 2H), 1.53 – 1.11 (m, J = 32.3, 22.8, 10.9 Hz, 4H).
ormation of N-[(IR, 3S>3-[[5-fluoro-2-(5-fluoro-1H-pyrrolo[2,3- b]pyridin-3-yl)pyrimidin-4-yl] amino] cyclohexyl]morpholine-4-carboxamide (706)
N- [( IR, 35)-3 – [ [5 -fluoro-2- [5 -fluoro- 1 -(p-tolylsulfonyl)pyrrolo [2,3 -b]pyridin-3 – yl]pyrimidin-4-yl]amino]cyclohexyl]morpholine-4-carboxamide, 44f, (2.0 g, 3.2 mmol) was suspended in methanol (50 mL) and treated with 25% sodium methoxide in methanol (19.9 mL, 92.3 mmol) . After stirring for 1 hour, the solvent was evaporated under reduced pressure, and the residue was partitioned between water (100 mL) and ethyl acetate (100 mL). The organic layer was collected, dried on Νa2SO4 and concentrated to provide the crude product as a yellow solid. This material was purified by silica gel chromatography on a 4Og column, using DCM/MeOH 1-6%. The purified fractions were treated with 2N HCl in ether and concentrated to provide 1.5 g of N-[(1R, 35)-3-[[5-fluoro-2-(5-fluoro-1H- pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl]amino]cyclohexyl]-morpholine-4-carboxamide as a white solid.
HCI D DCM
44e
Formation of (IS, S^-M-^-fluoro-S-CS-fluoro-1H-pyrrolo^S-^pyridin- 3-yl)phenyl)cyclohexane-1,3-diamine (44e)
To a solution of tert-butyl (IR, 35)-3-(2-fluoro-5-(5-fluoro-1-tosyl-lH-pyrrolo-[2,3- &]pyridin-3-yl)phenylamino)cyclohexylcarbamate, 44d, (0.65 g, 1.09 mmol) in methylene chloride (22 mL) was added hydrogen chloride (2.71 mL of 4M solution in 1,4-dioxane, 10.86 mmol). The reaction was heated to 50 °C and stirred for 6 hours. The mixture was cooled to room temperature and concentrated in vacuo, producing a yellow solid. The crude residue was purified via silica gel chromatography (25-50% Ethyl Acetate/hexanes gradient). Desired fractions were combined and concentrated in vacuo to produce 350 mg of 44e as a yellow powder.
General Scheme 67 SIMILAR TO A POINT BUT NOT SAME
(a) Pd/C (wet, Degussa), hydrogen, EtOH (b) 2,4-dichloro-5-fluoropyrimidine, 1Pr2NEt, THF, reflux (c) LiOH, THF/water, 5O°C
SIMILAR BUT NOT SAME
(d) DPPA, Et3N, THF, 85 °C (e) 5-fluoro-3-(4,4,5,5-tetramethyl-1,3 ,2-dioxaborolan-2-yl)-1- tosyl-l//-pyrrolo[2,3-i]pyridine, XPhos, Pd2(dba)3, K3PO4, 2-methylTHF, water, 125 °C (f)
Formation (IR, 35)-ethyl 3-aminocyclohexanecarboxylate (67b)
To a solution of (IR, 35)-ethyl 3-(benzyloxycarbonylamino)cyclohexane-carboxylate, 18b, (14.0 g, 45.9 mmol) in ethanol (3 mL) was added Pd/C (wet, Degussa (2.4 g, 2.3 mmol). The mixture was evacuated and then stirred under atmosphere of nitrogen at room temperature overnight. The reaction mixture was filtered through a pad of celite and the resulting filtrate concentrated in vacuo to provide an oil that was used without further purification.
Formation (IR, SS^-ethyl 3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexane- carboxylate (67c)
To a solution of (IR, 3«S)-ethyl S-aminocyclohexanecarboxylate, 67b, (5.1 g, 24.1 mmol) and 2,4-dichloro-5,-fluoropyrimidine (6.0 g, 36.0 mmol) in THF (60 mL) was added diisopropylethylamine (9.6 mL, 55.4 mmol). The mixture was heated to reflux overnight. The reaction was cooled to room temperature and concentrated in vacuo. The residue was diluted with water and extracted twice with ethyl acetate. The combined organic phases were dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (0-40% EtOAc/hexanes gradient) to provide 6.7 g of (IR, 35*)-ethyl 3-(2- chloro-5-fluoropyrimidin-4-ylamino)cyclohexane-carboxylate as a white solid: LCMS RT = 3.1 (M+H) 302.2.
Formation (IR, 35)-3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexanecarboxylic acid (67d)
To a solution of (IR, 35*)-ethyl 3-(2-chloro-5-fluoropyrimidin-4- ylamino)cyclohexane-carboxylate, 67c, (20.0 g, 66.3 mmol) in THF (150 mL) was added added a solution of LiOH hydrate (8.3 g, 198.8 mmol) in 100ml water. The reaction mixture was stirred at 50 °C overnight, To the reaction mixture was added HCl (16.6 mL of 12 M solution, 198.8 mmol) and EtOAc. The organic phase was washed with brine and dried over MgSO4 and the solvent was removed under reduced pressure to afford 17.5 g of product that was used without further purification: 1H NMR (300 MHz, CDC13) δ 7.91 (d, J = 2.7 Hz, 2H), 5.24 (d, J = 7.3 Hz, 2H), 4.19 – 4.03 (m, 3H), 3.84 – 3.68 (m, 3H), 2.59 (ddd, J= 11.5, 8.2, 3.6 Hz, 2H), 2.38 (d, J = 12.4 Hz, 2H), 2.08 (d, J = 9.6 Hz, 6H), 1.99 – 1.76 (m, 5H), 1.63 – 1.34 (m, 6H), 1.32 – 1.15 (m, 4H).
Formation N-((1R, 35)-3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexyl)- pyrrolidine-1-carboxamide (67e)
A solution of (IR, 35)-3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexane- carboxylic acid, 67d, (8.2 g, 30.0 mmol), (azido(phenoxy)phosphoryl)oxybenzene (9.7 mL, 45.0 mmol) and triethylamine (5.8 mL, 42.0 mmol) in THF (200 mL) was degassed under nitrogen for 15 minutes. The reaction mixture was heated at 85 °C for 30 minutes until LC/MS indicated complete consumption of carboxylic acid, 67d. To the reaction mixture was added pyrrolidine (7.5 mL, 90.0 mmol) and the reaction was heated at 85 °C for an additional 15 min. The mixture was diluted into brine and extracted with EtOAc. The organic phase was separated, dried over MgSO4. The product was isolated (6.25 g) by filtration after partial removal of solvent in vacuo: 1H NMR (300 MHz, CDC13) δ 7.87 (d, J = 2.8 Hz, 2H), 5.04 (d, J = 8.1 Hz, 2H), 4.09 (ddd, J = 26.9, 13.4, 5.6 Hz, 4H), 3.91 – 3.71 (m, 2H), 3.32 (t, J= 6.5 Hz, 7H), 2.45 (d, J= 11.5 Hz, 2H), 2.08 (dd, J= 22.1, 12.0 Hz, 4H), 1.96- 1.82 (m, 9H), 1.54 (dd, J= 18.6, 8.5 Hz, 2H), 1.22 – 1.01 (m, 6H).
Formation N-((IR, 3S>3-(5-fluoro-2-(5-fluoro-1-tosyl-1H-pyrrolo[2,3-b]pyridm-3- yl)pyrimidin-4-ylamino)cyclohexyl)pyrrolidine-1-carboxamide (67f)
A solution of N-((1R, 3«S)-3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexyl)- pyrrolidine-1-carboxamide, 67e, (6.8 g, 20.0 mmol), 5-fluoro-1-(p-tolylsulfonyl)-3-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)pyrrolo[2,3-b]pyridine, 44a, (12.5 g, 30.0 mmol) and K3PO4 (17.0 g, 80.0 mmol) in 2-methyl TΗF (180 mL) and water (20 mL) was degassed under nitrogen for 30 min. To the mixture was added dicyclohexyl-[2-(2,4,6- triisopropylphenyl)phenyl]phosphane (XPhos) (1.1 g, 2.4 mmol) and Pd2(dba)3 (0.5 g, 0.5 mmol). The reaction mixture was heated in a pressure bottle at 125 °C for 2.5 hr. The reaction mixture was filtered through celite, the solvent was removed under reduced pressure. The resulting residue was purified by silica gel chromatography (8%MeOΗ/CΗ2Cl2) to afford 11.5 g of the desired product: 1H ΝMR (300 MHz, CDC13) δ 8.54 (s, 1H), 8.49 (dd, J= 9.0, 2.8 Hz, 1H), 8.32 (d, J= 2.1 Hz, 1H), 8.13 (d, J= 8.3 Hz, 2H), 8.07 (d, J= 3.2 Hz, 1H), 7.30 (d, J = 8.5 Hz, 2H), 4.98 (d, J = 6.3 Hz, 1H), 4.37 – 4.16 (m, 1H), 4.08 (d, J = 7.3 Hz, 1H), 3.99 – 3.80 (m, 1H), 3.33 (t, J= 6.5 Hz, 4H), 2.52 (d, J= 11.6 Hz, 1H), 2.39 (s, 3H), 2.29 (d, J= 11.3 Hz, 1H), 2.12 (d, J= 11.1 Hz, 1H), 1.99 – 1.81 (m, 5H), 1.70 – 1.55 (m, 1H), 1.22 – 1.08 (m, 2H).
Formation N-((IR, 3S>3-(5-fluoro-2-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)- pyrimidin-4-ylamino)cyclohexyl)pyrrolidine-1-carboxamide (895)
A solution of N-((1R, 35)-3-(5-fluoro-2-(5-fluoro-1-tosyl-lH-pyrrolo[2,3-b]pyridin-3- yl)pyrimidin-4-ylamino)cyclohexyl)pyrrolidine-1-carboxamide, 67f, (11.5 g, 19.3 mmol) in TΗF (150 mL) was added sodium methoxide (4.173 g, 19.31 mmol). After stirring the reaction mixture for 2 minutes, the mixture was poured into an aqueous saturated solution of NaHCO3. The organic phase was washed with brine, dried over MgSO4 and the solvent was removed under reduced pressure. The resulting residue was purified by silica gel chromatography (10%MeOH/CH2Cl2) to afford 6.5g of the desired product. The product was converted to an HCl salt by dissolving in MeOH (100 mL) and adding 2.4 mL of 12M HCl solution at room temperature. The solution was stirred at for lhour and the HCl salt precipitated out and filtered to provide 7.05g of the HCl salt: 1H NMR (300 MHz, DMSO) δ 9.36 (s, 2H), 9.05 (d, J= 3.0 Hz, 2H), 8.49 (d, J= 5.6 Hz, 2H), 8.41 (dd, J= 2.6, 1.4 Hz, 2H), 8.31 (d, J= 9.5 Hz, 2H), 5.92 (s, 3H), 4.24 (s, 3H), 3.64 (s, 2H), 3.18 (t, J= 6.6 Hz, 7H), 2.07 (dt, J = 22.7, 11.5 Hz, 4H), 1.87 (t, J = 12.6 Hz, 4H), 1.77 (dd, J = 8.0, 5.3 Hz, 7H), 1.65 – 1.13 (m, 8H).
PATENT
US-20120171245-A1 / 2012-07-05
INHIBITORS OF INFLUENZA VIRUSES REPLICATION
/////////VX-? , an Azaindolyl-Pyrimidine Inhibitor, Influenza Virus Replication, Vertex, preclinical, 1259498-06-0
O=C(NC1CCC[C@@H](C1)Nc2nc(ncc2F)\C\4=C\N=C3\N\C=C(\F)/C=C3/4)N5CCCCC5
Filed under:
Preclinical drugs Tagged:
1259498-06-0,
an Azaindolyl-Pyrimidine Inhibitor,
Influenza Virus Replication,
preclinical,
VERTEX,
VX-? ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()























.jpg)











































































![CAS # 13103-34-9, Boldenone undecylenate, Boldenone undec-10-enoate, 17b-[(1-Oxo-10-undecenyl)oxy]-androsta-1,4-dien-3-one, 17b-Hydroxyandrosta-1,4-dien-3-one 10-undecenoate](http://i1.wp.com/www.chemblink.com/structures/13103-34-9.gif)


























































 .
.






















.jpg?n=3254)





















































.bmp)
.bmp)
.gif)