
Doravirine, MK-1439……….. AN ANTIVIRAL
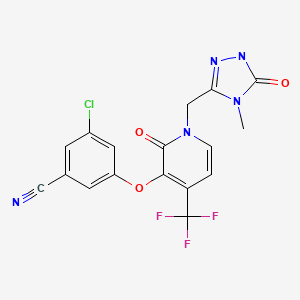
3-Chloro-5-({1-[(4-methyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)methyl]-2-oxo-4-(trifluoromethyl)-1,2-dihydro-3-pyridinyl}oxy)benzonitrile
Benzonitrile, 3-chloro-5-[[1-[(4,5-dihydro-4-methyl-5-oxo-1H-1,2,4-triazol-3-
yl)methyl]-1,2-dihydro-2-oxo-4-(trifluoromethyl)-3-pyridinyl]oxy]-
3-chloro-5-({1-[(4-methyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)methyl]-2-
oxo-4-(trifluoromethyl)-1,2-dihydropyridin-3-yl}oxy)benzonitrile
1338225-97-0 CAS
MOLECULAR FORMULA C17H11ClF3N5O3
MOLECULAR WEIGHT 425.7
Merck Sharp & Dohme Corp
reverse transcriptase inhibitor
Doravirine (MK-1439) is a non-nucleoside reverse transcriptase inhibitor under development by Merck & Co. for use in the treatment of HIV infection. Doravirine demonstrated robust antiviral activity and good tolerability in a small clinical study of 7-day monotherapy reported at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013. Doravirine appeared safe and generally well tolerated with most adverse events being mild-to-moderate.[1][2]
investigational next-generation, non-nucleoside reverse transcriptase inhibitor (NNRTI), at the 21st Conference on Retroviruses and Opportunistic Infections (CROI). Interim data demonstrating potent antiretroviral (ARV) activity for four doses (25, 50, 100 and 200 mg) of once-daily, oral doravirine in combination with tenofovir/emtricitabine in treatment-naïve, HIV-1 infected adults after 24 weeks of treatment were presented during a late-breaker oral session. Based on these findings as well as other data from the doravirine clinical program, Merck plans to initiate a Phase 3 clinical trial program for doravirine in combination with ARV therapy in the second half of 2014.
“Building on our long-standing commitment to the HIV community, Merck continues to evaluate new candidates we believe have the potential to make a meaningful difference in the lives of HIV patients,” said Daria Hazuda, Ph.D., vice president, Infectious Diseases, Merck Research Laboratories. “We look forward to advancing doravirine into Phase 3 clinical trials in the second half of 2014.”
Doravirine Clinical Data
This randomized, double-blind clinical trial examined the safety, tolerability and efficacy of once-daily doravirine (25, 50, 100 and 200 mg) in combination with once-daily tenofovir/emtricitabine versus efavirenz (600 mg), in treatment-naïve, HIV-1 infected patients. The primary efficacy analysis was percentage of patients achieving virologic response (< 40 copies/mL).
At 24 weeks, doravirine doses of 25, 50, 100, and 200 mg showed virologic response rates consistent with those observed for efavirenz at a dose of 600 mg. All treatment groups showed increased CD4 cell counts.
|
Proportion of Patients with Virologic |
Mean CD4 Change |
|||||||
| Treatment* | Dose (mg) | n/N |
% <40 |
cells/μL
|
||||
| Doravirine | 25 | 32/40 | 80.0 (64.6, 90.9) | 158 (119, 197) | ||||
| 50 | 32/42 | 76.2 (60.5, 87.9) | 116 (77, 155) | |||||
| 100 | 30/42 | 71.4 (55.4, 84.3) | 134 (100, 167) | |||||
| 200 | 32/41 | 78.0 (62.4, 89.4) | 141 (96, 186) | |||||
| Efavirenz | 600 | 27/42 | 64.3 (48.0, 78.4) | 121 (73, 169) | ||||
| Missing data approach: | Non-completer = Failure | Observed Failure | ||||||
|
*In combination with tenofovir/emtricitabine |
||||||||
The incidence of drug-related adverse events was comparable among the doravirine-treated groups. The overall incidence of drug-related adverse events was lower in the doravirine-treated groups (n=166) than the efavirenz-treated group (n=42), 35 percent and 57 percent, respectively. The most common central nervous system (CNS) adverse events at week 8, the primary time point for evaluation of CNS adverse experiences, were dizziness [3.0% doravirine (overall) and 23.8% efavirenz], nightmare [1.2% doravirine (overall) and 9.5% efavirenz], abnormal dreams [9.0% doravirine (overall) and 7.1% efavirenz], and insomnia [5.4% doravirine (overall) and 7.1% efavirenz].
Based on the 24-week data from this dose-finding study, a single dose of 100 mg doravirine was chosen to be studied for the remainder of this study, up to 96 weeks.
About Doravirine
DORAVIRINE
Doravirine, also known as MK-1439, is an investigational next-generation, NNRTI being evaluated by Merck for the treatment of HIV-1 infection. In preclinical studies, doravirine demonstrated potent antiviral activity against HIV-1 with a characteristic profile of resistance mutations selected in vitro compared with currently available NNRTIs. In early clinical studies, doravirine demonstrated a pharmacokinetic profile supportive of once-daily dosing and did not show a significant food effect.
Merck’s Commitment to HIV
For more than 25 years, Merck has been at the forefront of the response to the HIV epidemic, and has helped to make a difference through our proud legacy of commitment to innovation, collaborating with the community, and expanding global access to medicines. Merck is dedicated to applying our scientific expertise, resources and global reach to deliver healthcare solutions that support people living with HIV worldwide.

About Merck

Today’s Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside the United States and Canada. Through our prescription medicines, vaccines, biologic therapies, and consumer care and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit www.merck.com and connect with us on Twitter, Facebook and YouTube.
Discovery of MK-1439, an orally bioavailable non-nucleoside reverse transcriptase inhibitor potent against a wide range of resistant mutant HIV viruses
Bioorg Med Chem Lett 2014, 24(3): 917
http://www.sciencedirect.com/science/article/pii/S0960894X13014546
The optimization of a novel series of non-nucleoside reverse transcriptase inhibitors (NNRTI) led to the identification of pyridone 36. In cell cultures, this new NNRTI shows a superior potency profile against a range of wild type and clinically relevant, resistant mutant HIV viruses. The overall favorable preclinical pharmacokinetic profile of 36 led to the prediction of a once daily low dose regimen in human. NNRTI 36, now known as MK-1439, is currently in clinical development for the treatment of HIV infection.


Scheme 1.
Reagents and conditions: (a) K2CO3, NMP, 120 °C; (b) KOH, tert-BuOH, 75 °C; (c) Zn(CN)2, Pd(PPh3)4, DMF, 100 °C.

Reagents and conditions: (a) K2CO3, DMF, −10 °C; (b) MeI or EtI, K2CO3, DMF.
36 IS DORAVIRINE
WO 2011120133
http://www.google.com/patents/WO2011120133A1?cl=en
Scheme I depicts a method for preparing compounds of Formula I in which hydroxypyridine 1-1 is alkylated with chlorotriazolinone 1-2 to provide 1-3 which can be selectively alkylated with an alkyl halide (e.g., methyl iodide, ethyl iodide, etc.) to afford the desired 1-4. Scheme I

Scheme II depicts an alternative route to compounds of the present invention, wherein fluorohydroxypyridine II-l can be alkylated with chlorotriazolinone II-2 to provide the alkylated product II-3 which can be converted to the desired II-5 via nucleophilic aromatic substitution (S] fAr) using a suitable hydroxyarene II-4.
Scheme II

Hydroxypyridines of formula I-l (Scheme 1) can be prepared in accordance with Scheme III, wherein a SNAr reaction between pyridine III-l (such as commercially available 2- chloro-3-fluoro-4-(trifluoromethyl)pyridine) and hydroxyarene H-4 can provide chloropyridine III-2, which can be hydrolyzed under basic conditions to the hydroxypyridine I-l. Scheme III

Another method for preparing hydroxypyridines of formula I-l is exemplified in Scheme IV, wherein S Ar coupling of commercially available 2-chloro-3-fluoro-4- nitropyridone-N-oxide IV-1 with a suitable hydroxyarene II-4 provides N-oxide IV-2, which can first be converted to dihalides IV-3 and then hydro lyzed to hydroxypyridine IV-4. Further derivatization of hydroxypyridine IV-4 is possible through transition metal-catalyzed coupling processes, such as Stille or boronic acid couplings using a PdLn catalyst (wherein L is a ligand such as triphenylphosphine, tri-tert-butylphosphine or xantphos) to form hydroxypyridines IV-5, or amination chemistry to form hydroxypyridines IV-6 in which R2 is N(RA)RB.
Scheme IV

IV-1

- – Scheme V depicts the introduction of substitution at the five-position of the hydroxypyridines via bromination, and subsequent transition metal-catalyzed chemistries, such as Stille or boronic acid couplings using PdLn in which L is as defined in Scheme IV to form hydroxypyridines V-3, or amination chemistry to form hydroxypyridines V-4 in which R3 is N(RA)RB.
Scheme V

As shown in Scheme IV, fiuorohydroxypyridines II-l (Scheme II) are available from the commercially available 3-fluoroypridines VI- 1 through N-oxide formation and rearrangement as described in Konno et al., Heterocycles 1986, vol. 24, p. 2169.
Scheme VI

The following examples serve only to illustrate the invention and its practice. The examples are not to be construed as limitations on the scope or spirit of the invention.
The term “room temperature” in the examples refers to the ambient temperature which was typically in the range of about 20°C to about 26°C.
EXAMPLE 1
3-Chloro-5-({ l-[(4-methyl-5-oxo-4,5-dihydro-lH-l ,2,4-triazol-3-yl)methyl]-2-oxo-4- (trifluoromethyl)-l ,2-dihydropyridin-3-yl}oxy)benzonitrile (1-1)

Step 1(a):

A mixture of the 3-bromo-5-chlorophenol (3.74 g; 18.0 mmol), 2-chloro-3-fluoro- 4-(trifluoromethyl)pyridine (3.00 g; 15.0 mmol) and 2CO3 (2.49 g; 18.0 mmol) in NMP (15 mL) was heated to 120°C for one hour, then cooled to room temperature. The mixture was then diluted with 250 mL EtOAc and washed with 3 x 250 mL 1 :1 H20:brine. The organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (120 g column; load with toluene; 100:0 to 0:100 hexanes:CH2Cl2 over 40 minutes) provided title compound (1-2) as a white solid. Repurification of the mixed fractions provided additional title compound. lH NMR (400 MHz, CDCI3): δ 8.55 (d, J = 5.0 Hz, 1 H); 7.64 (d, J = 5.0 Hz, 1 H);
7.30 (s, 1 H); 6.88 (s, 1 H); 6.77 (s, 1 H).
3-(3-bromo-5-chlorophenoxy)-4-(trifluoromethyl)pyridin-2-ol (1-3)

To a suspension of 3-(3-bromo-5-chlorophenoxy)-2-chloro-4- (trifluoromethyl)pyridine (1-2; 3.48 g; 8.99 mmol) in lBuOH (36 mL) was added KOH (1.51 g; 27.0 mmol) and the mixture was heated to 75°C overnight, at which point a yellow oily solid had precipitated from solution, and LCMS analysis indicated complete conversion. The mixture was cooled to room temperature, and neutralized by the addition of -50 mL saturated aqueous NH4CI. The mixture was diluted with 50 mL H2O, then extracted with 2 x 100 mL EtOAc. The combined organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (120 g column; dry load; 100:0 to 90: 10 CH2Cl2:MeOH over 40 minutes) provided the title compound (1-3) as a fluffy white solid. lH NMR (400 MHz, DMSO): δ 12.69 (s, 1 H); 7.59 (d, J = 6.9 Hz, 1 H); 7.43 (t, J = 1.7 Hz, 1 H); 7.20 (t, J = 1.9 Hz, 1 H); 7.13 (t, J = 2.0 Hz, 1 H); 6.48 (d, J = 6.9 Hz, 1 H).
3-chloro-5-{[2-hydroxy-4-(trifluoromethyl)pyridin-3-yl]oxy}benzonitrile (1-4)

To a suspension of 3-(3-bromo-5-chlorophenoxy)-4-(trifluoromethyl)pyridin-2-ol (1-3; 3.25 g; 8.82 mmol) in NMP (29 mL) was added CuCN (7.90 g; 88 mmol) and the mixture was heated to 175°C for 5 hours, then cooled to room temperature slowly. With increased fumehood ventilation, 100 mL glacial AcOH was added, then 100 mL EtOAc and the mixture was filtered through Celite (EtOAc rinse). The filtrate was washed with 3 x 200 mL 1 : 1 H20:brine, then the organic extracts were dried (Na2S04) and concentrated in vacuo.
Purification by ISCO CombiFlash (120 g column; dry load; 100:0 to 90:10 CH2Cl2:MeOH over 40 minutes), then trituration of the derived solid with Et20 (to remove residual NMP which had co-eluted with the product) provided the title compound (1-4). lH NMR (400 MHz, DMSO): δ 12.71 (s, 1 H); 7.75 (s, 1 H); 7.63-7.57 (m, 2 H); 7.54 (s, 1 H); 6.49 (d, J = 6.9 Hz, 1 H).
Step 1(d): 5-(chloromethyl)-2,4-dihydro-3H-l,2,4-triazol-3-one (1-5)

The title compound was prepared as described in the literature: Cowden, C. J.; Wilson, R. D.; Bishop, B. C; Cottrell, I. F.; Davies, A. J.; Dolling, U.-H. Tetrahedron Lett. 2000, 47, 8661.
3 -chloro-5 -( { 2-oxo- 1 – [(5 -oxo-4,5 -dihydro- 1 H- 1 ,2,4-triazol-3 -yl)methyl] – 4- (trifiuoromethyl)- 1 ,2-dihydropyridin-3 -yl } oxy)benzonitrile (1-6)

A suspension of the 3-chloro-5-{[2-hydroxy-4-(trifluoromethyl)pyridin-3- yl]oxy}benzonitrile (1-4; 2.00 g; 6.36 mmol), 5-(chloromethyl)-2,4-dihydro-3H-l,2,4-triazol-3- one (1-5; 0.849 g; 6.36 mmol) and K2CO3 (0.878 g; 6.36 mmol) in DMF (32 mL) was stirred for 2 hours at room temperature, at which point LCMS analysis indicated complete conversion. The mixture was diluted with 200 mL Me-THF and washed with 150 mL 1 : 1 : 1 H20:brine:saturated aqueous NH4CI, then further washed with 2 x 150 mL 1 : 1 H20:brine. The aqueous fractions were further extracted with 150 mL Me-THF, then the combined organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (80 g column; dry load; 100:0 to 90:10 EtOAc:EtOH over 25 minutes) provided the title compound (1-6) as a white solid. lH NMR (400 MHz, DMSO): δ 1 1.46 (s, 1 H); 1 1.39 (s, 1 H); 7.93 (d, J = 7.3 Hz, 1 H); 7.76 (s, 1 H); 7.58 (s, 1 H); 7.51 (s, 1 H); 6.67 (d, J = 7.3 Hz, 1 H); 5.02 (s, 2 H).
Step 1(f): 3 -chloro-5 -( { 1 – [(4-methyl-5-oxo-4,5 -dihydro- 1 H- 1 ,2,4-triazol-3 -yl)methyl] -2- oxo-4-(trifluoromethyl)- 1 ,2-dihydropyridin-3 -yl } oxy)benzonitrile (1 -1 )
A solution of 3-chloro-5-({2-oxo-l -[(5-oxo-4,5-dihydro-lH-l,2,4-triazol-3- yl)methyl]- 4-(trifluoromethyl)-l ,2-dihydropyridin-3-yl}oxy)benzonitrile (1-6; 2.37 g; 5.76 mmol) and K2CO3 (0.796 g; 5.76 mmol) in DMF (58 mL) was cooled to 0°C, then methyl iodide (0.360 mL; 5.76 mmol) was added. The mixture was allowed to warm to room
temperature, and stirred for 90 minutes, at which point LCMS analysis indicated >95%
conversion, and the desired product of -75% LCAP purity, with the remainder being unreacted starting material and 6/s-methylation products. The mixture was diluted with 200 mL Me-THF, and washed with 3 x 200 mL 1 : 1 H20:brine. The aqueous fractions were further extracted with 200 mL Me-THF, then the combined organic extracts were dried (Na2S04) and concentrated in vacuo. The resulting white solid was first triturated with 100 mL EtOAc, then with 50 mL THF, which provided (after drying) the title compound (1-1) of >95% LCAP. Purification to >99% LCAP is possible using Prep LCMS (Max-RP, 100 x 30 mm column; 30-60% CH3CN in 0.6% aqueous HCOOH over 8.3 min; 25 mL/min). lH NMR (400 MHz, DMSO): δ 1 1.69 (s, 1 H); 7.88 (d, J = 7.3 Hz, 1 H); 7.75 (s, 1 H); 7.62 (s, 1 H); 7.54 (s, 1 H); 6.67 (d, J = 7.3 Hz, 1 H); 5.17 (s, 2 H); 3.1 1 (s, 3 H). EXAMPLE 1A
3-Chloro-5-({ l-[(4-methyl-5-oxo-4,5-dihydro-lH-l ,2,4-triazol-3-yl)methyl]-2- (trifluoromethyl)-l ,2-dihydropyridin-3-yl}oxy)benzonitrile (1-1)

Step lA(a): 2-chloro-3-(3-chloro-5-iodophenoxy)-4-(trifluoromethyl)pyridine (1A-2)

A mixture of the 3-chloro-l-iodophenol (208 g; 816.0 mmol), 2-chloro-3-fluoro-
4-(trifluoromethyl)pyridine (155 g; 777.0 mmol) and K2CO3 (161 g; 1 165.0 mmol) in NMP (1.5 L) was held at 60°C for 2.5 hours, and then left at room temperature for 2 days. The mixture was then re-heated to 60°C for 3 hours, then cooled to room temperature. The mixture was then diluted with 4 L EtOAc and washed with 2 L water + 1 L brine. The combined organics were then washed 2x with 500 mL half brine then 500 mL brine, dried over MgS04 and concentrated to afford crude 1A-2. lH NMR (500 MHz, DMSO) δ 8.67 (d, J = 5.0 Hz, 1 H), 7.98 (d, J = 5.0 Hz, 1 H), 7.63-7.62 (m, 1 H), 7.42-7.40 (m, 1 H), 7.22 (t, J = 2.1 Hz, 1 H).
Step lA(b): 2-chloro-3-(3-chloro-5-iodophenoxy)-4-(trifluoromethyl)pyridine (1A-3)

To a suspension of 3-(3-chloro-5-iodophenoxy)-2-chloro-4- (trifluoromethyl)pyridine (1A-2; 421 g, 970 mmol) in t-BuOH (1 L) was added KOH (272 g, 4850 mmol) and the mixture was heated to 75°C for 1 hour, at which point HPLC analysis indicated >95% conversion. The t-BuOH was evaporated and the mixture diluted with water (7mL/g, 2.4L) and then cooled to 0°C, after which 12N HC1 (~240mL) was added until pH 5. This mixture was then extracted with EtOAc (20mL/g, 6.5L), back extracted with EtOAc 1 x 5mL/g (1.5L), washed 1 x water:brine 1 : 1 (l OmL/g, 3.2L), 1 x brine (lOmL/g, 3.2L), dried over MgS04, filtered and concentrated to afford a crude proudct. The crude product was suspended in MTBE (2.25 L, 7mL/g), after which hexanes (1 L, 3 mL/g) was added to the suspension over ten minutes, and the mixturen was aged 30minutes at room temperature. The product was filtered on a Buchner, rinsed with MTBE hexanes 1 :2 (2 mL/g = 640 mL), then hexanes
(640mL), and dried on frit to afford 1A-3. lH NMR (400 MHz, acetone-d6): δ 11.52 (s, 1 H); 7.63 (d, J = 7.01 Hz, 1 H); 7.50-7.48 (m, 1 H); 7.34-7.32 (m, 1 H); 7.09-7.07 (m, 1 H); 6.48 (d, J = 7.01 Hz, 1 H).
Step lA(c): 3-chloro-5-{[2-hydroxy-4-(trifluoromethyl)pyridin-3-yl]oxy}benzonitrile (1-4)

A solution of 3-(3-chloro-5-iodophenoxy)-4-(trifluoromethyl)pyridin-2-ol (1A-3; 190 g; 457 mmol) in DMF (914 mL) was degassed for 20 minutes by bubbling N2, after which CuCN (73.7 g; 823 mmol) was added, and then the mixture was degassed an additional 5 minutes. The mixture was then heated to 120°C for 17 hours, then cooled to room temperature and partitioned between 6 L MeTHF and 2 L ammonium buffer (4:3: 1 = NH4CI
sat/water/NH-iOH 30%). The organic layer washed with 2 L buffer, 1 L buffer and 1 L brine then, dried over MgS04 and concentrated. The crude solid was then stirred in 2.2 L of refluxing
MeCN for 45 minutes, then cooled in a bath to room temperature over 1 hour, aged 30 minutes, then filtered and rinsed with cold MeCN (2 x 400mL). The solid was dried on frit under N2 atm for 60 hours to afford title compound 1-4. lH NMR (400 MHz, DMSO): δ 12.71 (s, 1 H); 7.75 (s, 1 H); 7.63-7.57 (m, 2 H); 7.54 (s, 1 H); 6.49 (d, J = 6.9 Hz, 1 H).
Steps lA(d) and lA(e)
The title compound 1-1 was then prepared from compound 1-4 using procedures similar to those described in Steps 1(d) and 1(e) set forth above in Example 1.
New patent
Crystalline anhydrous Form II of doravirine, useful for the treatment of HIV-1 and HIV-2 infections. The compound was originally claimed in WO2008076223. Also see WO2011120133. Merck & Co is developing doravirine (MK-1439), for the oral tablet treatment of HIV-1 infection. As of April 2014, the drug is in Phase 2 trials.
References
- Safety and Antiviral Activity of MK-1439, a Novel NNRTI, in Treatment-naïve HIV+ Patients. Gathe, Joseph et al. 20th Conference on Retroviruses and Opportunistic Infections. 3–6 March 2013. Abstract 100.
- CROI 2013: MK-1439, a Novel HIV NNRTI, Shows Promise in Early Clinical Trials. Highleyman, Liz. HIVandHepatitis.com. 6 March 2013.
The next-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) doravirine (formerly MK-1439) showed potent antiretroviral activity and good tolerability in combination with tenofovir/FTC (the drugs in Truvada) in a dose-finding study presented at the 21st Conference on Retroviruses and Opportunistic Infections (CROI) last week in Boston.
NNRTIs are generally well tolerated and well suited for first-line HIV treatment, but as a class they are susceptible to resistance. Pre-clinical studies showed that Merck’s doravirine has a distinct resistance profile and remains active against HIV with common NNRTI resistance mutations including K103N and Y181C.
As reported at last year’s CROI, doravirine reduced HIV viral load by about 1.3 log in a seven-day monotherapy study. Doravirine is processed by the CYP3A4 enzyme, but it is neither a CYP3A4 inducer nor inhibitor, so it is not expected to have major drug interaction concerns.
Javier Morales-Ramirez from Clinical Research Puerto Rico reported late-breaking findings from a phase 2b study evaluating the safety and efficacy of various doses of doravirine versus efavirenz (Sustiva) for initial antiretroviral therapy.
This study included 208 treatment-naive people living with HIV from North America, Europe and Asia. More than 90% were men, 74% were white, 20% were black and the median age was 35 years. At baseline, the median CD4 cell count was approximately 375 cells/mm3 and 13% had received an AIDS diagnosis. Study participants were stratified by whether their viral load was above (about 30%) or below 100,000 copies/ml; median HIV RNA was approximately 4.5 log10.
Morales-Ramirez reported 24-week results from part 1 of the study, which will continue for a total of 96 weeks. In this part, participants were randomly allocated into five equal-sized arms receiving doravirine at doses of 25, 50, 100 or 200mg once daily, or else efavirenz once daily, all in combination with tenofovir/FTC.
At 24 weeks, 76.4% of participants taking doravirine had viral load below 40 copies/ml compared with 64.3% of people taking efavirenz. Response rates were similar across doravirine doses (25mg: 80.0%; 50mg: 76.2%; 100mg: 71.4%; 200mg: 78.0%). More than 80% of participants in all treatment arms reached the less stringent virological response threshold of <200 copies/ml.
Both doravirine and efavirenz worked better for people with lower pre-treatment viral load in an ad hoc analysis. For people with <100,000 copies/ml at baseline, response rates (<40 copies/ml) ranged from 83 to 89% with doravirine compared with 74% with efavirenz. For those with >100,000 copies/ml, response rates ranged from 50 to 91% with doravirine vs 54% with efavirenz.
Median CD4 cell gains were 137 cells/mm3 for all doravirine arms combined and 121 cells/mm3 for the efavirenz arm.
Doravirine was generally safe and well tolerated. People taking doravirine were less than half as likely as people taking efavirenz to experience serious adverse events (3.0% across all doravirine arms vs 7.1% with efavirenz) or to stop treatment for this reason (2.4 vs 4.8%). Four people taking doravirine and two people taking efavirenz discontinued due to adverse events considered to be drug-related.
The most common side-effects were dizziness (3.6% with doravirine vs 23.8% with efavirenz), abnormal dreams (9.0 vs 7.1%), diarrhoea (4.8 vs 9.5%), nausea (7.8 vs 2.4%) and fatigue (6.6 vs 4.8%). Other central nervous system (CNS) adverse events of interest included insomnia (5.4 vs 7.1%), nightmares (1.2 vs 9.5%) and hallucinations (0.6 vs 2.4%). Overall, 20.5% of people taking doravirine reported at least one CNS side-effect, compared with 33.3% of people taking efavirenz.
People taking doravirine had more favourable lipid profiles and less frequent liver enzyme (ALT and AST) elevations compared with people taking efavirenz.
The researchers concluded that doravirine demonstrated potent antiretroviral activity in treatment-naive patients, a favourable safety and tolerability profile, and fewer drug-related adverse events compared with efavirenz.
Based on these findings, the 100mg once-daily dose was selected for future development and will be used in part 2 of this study, a dose-confirmation analysis that will enrol an additional 120 participants.
In the discussion following the presentation, Daniel Kuritzkes from Harvard Medical School noted that sometimes it takes longer for viral load to go down in people who start with a high level, so with further follow-up past 24 weeks doravirine may no longer look less effective in such individuals.
Reference
Morales-Ramirez J et al. Safety and antiviral effect of MK-1439, a novel NNRTI (+FTC/TDF) in ART-naive HIV-infected patients. 21st Conference on Retroviruses and Opportunistic Infections, Boston, abstract 92LB, 2014.
Merck Moves Doravirine Into Phase 3 Clinical Trials
Wednesday Mar 19 | Posted by: roboblogger | Full story: EDGE![]()
Earlier this month, at the 21st Conference on Retroviruses and Opportunistic Infections , Merck indicated plans to initiate a Phase 3 clinical trial program for doravirine in combination with ARV therapy in the second half of 2014.
Filed under: Phase3 drugs, Uncategorized Tagged: doravirine, MK 1439







