

Olaparib
オラパリブ
奥拉帕尼
Women suffering from advanced relapsed BRCA-mutated ovarian cancer could gain access to a new treatment option after European regulators waved through AstraZeneca’s Lynparza (olaparib).
The European Commission has approved the first-in-class PARP inhibitor for the maintenance treatment of adults with platinum-sensitive relapsed BRCA-mutated high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in complete response or partial response to platinum-based chemotherapy.

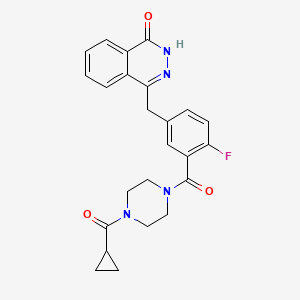
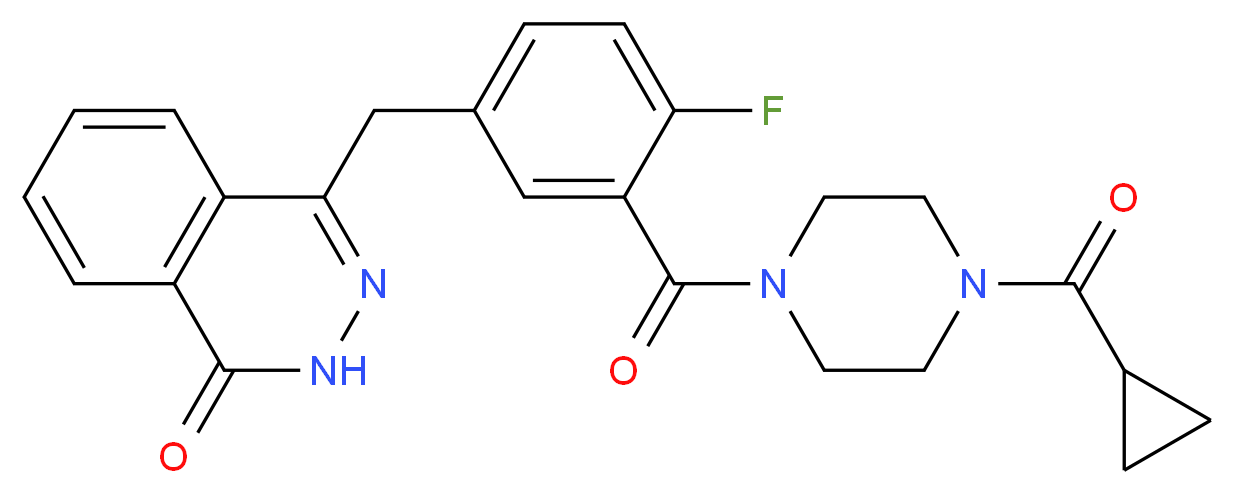
4-[[3-[4-(cyclopropanecarbonyl)piperazine-1-carbonyl]-4-fluorophenyl]methyl]-2H-phthalazin-1-one, cas 763113-22-0
KU-0059436
KU-59436
Olaparib (AZD-2281, trade name Lynparza) is an experimental chemotherapeutic agent, developed by KuDOS Pharmaceuticalsand later by AstraZeneca, that is currently undergoing clinical trials. It is an inhibitor of poly ADP ribose polymerase (PARP), an enzyme involved in DNA repair.[1] It acts against cancers in people with hereditary BRCA1 or BRCA2 mutations, which includes many ovarian, breast and prostate cancers.
Olaparib is an oral poly-ADP-ribose polymerase (PARP) enzyme inhibitor developed by AstraZeneca. The product is awaiting registration in the E.U. and US as a maintenance treatment of patients with BRCA mutated platinum-sensitive relapsed serous ovarian cancer. In 2014, positive opinion was received in the E.U. recommending Lynparza approval for the maintanance treatment of BRCA mutated platinum-sensitive relapsed serous ovarian cancer.
An oral poly (ADP ribose) polymerase (PARP) inhibitor being investigated by British drug company AstraZeneca, is seeking approval from the U.S. Food and Drug Administration (FDA) for the treatment of BRCA mutated platinum-sensitive relapsed ovarian cancer. AstraZeneca filed the US regulatory submission for olaparib in February 2014. Olaparib, one of several cancer drugs AstraZeneca flagged as having strong potential in its defense of a $118 billion take-over bid by Pfizer,was accepted for priority review on April 30, 2014 by the U.S. Food and Drug Administration (FDA). The NDA filing was based on Phase II study 19 data, a randomized, double-blind, placebo-controlled, Phase II study.
On June 25, 2014, FDA Oncologic Drugs Advisory Committee (ODAC), an advisory panel to the U.S. Food and Drug Administration (FDA), voted 11 to two against the accelerated approval of the PARP inhibitor olaparib as a maintenance therapy for women with platinum-sensitive relapsed ovarian cancer who have the germline BRCA (gBRCA) mutation, and who are in complete or partial response to platinum-based chemotherapy. By voting no, the committee recommended waiting for results from the larger confirmatory phase III SOLO-2 trial, which began enrolling in September 2013. According to clincialtrials.gov, the SOLO-2 study (NCT01874353) is slated to wrap in July 2015.
In terms of clinical development, phase III trials are ongoing at AstraZeneca for the treatment of gastric cancer and metastatic breast cancer. Olaparib is also in phase II clinical studies for several indications, including breast cancer, pancreatic cancer and castration-resistant prostate cancer. In March 2014, a phase II was also initiated in GB for the treatment of patients with stage IIIB or stage IV NSCLC that is not amenable to curative therapy. A phase I clinical trial for the treatment of melanoma has been completed. Phase II clinical trials are ongoing at General Hospital Corp. for the treatment of sarcoma. The drug had been in phase II clinical trials for the treatment of colorectal cancer; however no recent developments have been reported.
Discovered by KuDOS Pharmaceuticals, has experienced several twists and turns during its clinical development. Promising results for the drug were reported at the 2011 ASCO Annual Meeting, based on impressive early phase II results, only to have clinical development discontinued later that year after disappointing phase II trial results in a more generalized group of ovarian cancer patients. However, a re-analysis of the data in BRCA-positive patients – coupled with a reformulation of the drug – convinced the British drugmaker to think again and keep it going. AstraZeneca initiates Phase III clinical studies (SOLO 1 and SOLO 2) for olaparib in the U.S. in September 2013. AstraZeneca has filed Marketing Authorisation Application (MAA) for olaparib in EU in September 2013 based on Phase II study 19 data. The U.S. Food and Drug Administration has already granted olaparib orphan drug status for ovarian cancer and will hold an advisory panel hearing on the company’s application on June 25, 2014.
In 2013, orphan drug designation in the U.S. was assigned to the compound for the treatment of ovarian cancer. The compound was originally developed by Kudos Pharmaceuticals, which was acquired by AstraZeneca in 2006.
Early Phase I trials were promising, and olaparib underwent Phase II trials. However, in December 2011, AstraZeneca announced following interim analysis of a phase-II study which indicated that the previously reported progression free survival benefit was unlikely to translate into an overall survival benefit, that it would not progress into Phase III development for the maintenance treatment of serous ovarian cancer,[2] and took a charge of $285 million. The decision to discontinue development of the drug was reversed in 2013,[3] with AstraZeneca posting a new Phase III trial of Olaparib for patients with BRCA mutated ovarian cancer in April 2013.[4]
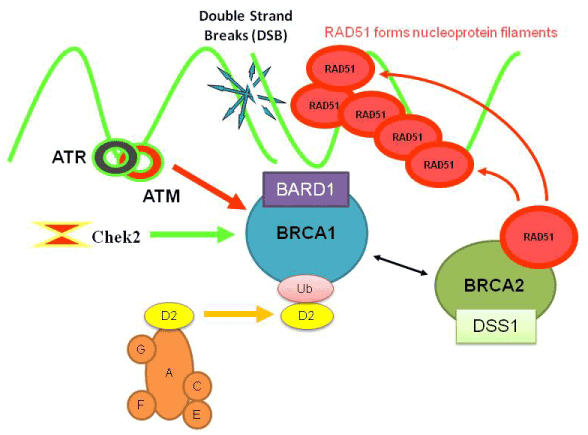
Mechanism of action
Olaparib acts as an inhibitor of the enzyme Poly ADP ribose polymerase (PARP) and is one of the first PARP inhibitors. Patients with BRCA1/2 mutations may be genetically predisposed to developing some forms of cancer, and are often resistant to other forms of cancer treatment, but this also sometimes gives their cancers a unique vulnerability, as the cancer cells have increased reliance on PARP to repair their DNA and enable them to continue dividing. This means that drugs which selectively inhibit PARP may be of significant benefit in patients whose cancers are susceptible to this treatment.[5][6][7][8][9][10]
Trial results
Phase I clinical trials, in patients with BRCA-mutated tumors including ovarian cancer, were encouraging.[11] In one of these studies, it was given to 19 patients with inherited forms of advanced breast, ovarian and prostate cancers caused by mutations of the BRCA1 and BRCA2 genes. In 12 of the patients, none of whom had responded to other therapies, tumours shrank or stabilised.[12] One of the first patients to be given the treatment (who had castration-resistant prostate cancer) was as of July 2009 still in remission after two years.
In 2009 Phase II clinical trials examining the efficacy of Olaparib in treating breast, ovarian and colorectal cancer were initiated.[13][14] A phase II trial that included 63 cases of ovarian cancer concluded that olaparib is promising for women with ovarian cancer. [7 responses in 17 patients with BRCA1 or BRCA2 mutations and 11 responses in the 46 who did not have these mutations.][15]
Side effects
Olaparib is generally well tolerated, the side effects consist mainly of fatigue, somnolence, nausea, loss of appetite and thrombocytopenia.

………………………

…………….
| LOU Xi-yu, YANG Xuan, DING Yi-li, WANG Jian-jun, YAN Qing-yan, HUANG Xian-gui, GUO Yang-hui, WANG Xiang-jing, XIANG Wen-sheng Synthesis of Olaparib Derivatives and Their Antitumor Activities  |
| 2013 Vol. 29 (2): 231-235 [摘要] ( 390 ) [HTML 1KB] [PDF 0KB] ( 22 ) doi: 10.1007/s40242-013-2448-5 |
……………………….

…………………
4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1
J Med Chem 2008, 51(20): 6581
…………………………..
http://www.google.co.in/patents/WO2004080976A1?cl=en
Synthesis of Key Intermediates
3- (4-0x0-3 , 4-dihydrophthalazin-l -ylmethyl) benzoic a cid (A)

A mixture of 27% sodium methoxide solution in methanol (400 g, 2 mol) and methanol (150 ml) was added dropwise between ambient temperature and 30°C over 15 minutes to a stirred mixture of phthalide (67 g, 0.5 mol), 3-formylbenzonitrile (65.5 g, 0.5 mol) and ethyl propionate (250 ml) , the mixture was stirred at ambient temperature for 40 minutes and at reflux temperature for 1 hour, then it was allowed to cool to ambient temperature. The resulting red solid was collected by filtration, washed with ethyl acetate (2 x 50 ml) and dissolved in water (1800 ml) . The solution was acidified by the addition of acetic acid (60 ml) and the resulting red solid was collected by filtration, washed with water (2 x 200 ml) and dried in vacuo to give 3- (1,3- dioxoindan-2-yl) benzonitrile (83.2 g) as a dark red solid, m.pt. 179- 182°C, m/z (M+H)+‘ 248, which was used without further purification.
3- (1, 3-Dioxoindan-2-yl) benzonitrile (74.18 g, 0.3 mol) was added in portions to a solution of sodium hydroxide (36 g, 0.9 mol) in water (580 ml), the resulting dark red suspension was stirred at reflux temperature for 5 hours, then it was cooled to ambient temperature and washed with ethyl acetate (3 x 300 ml) . The aqueous solution was acidified by the dropwise addition of concentrated hydrochloric acid (110 ml), the mixture was stirred at ambient temperature for 1 hour, then the resulting solid was collected by filtration, washed with water (2 x 200 ml) and dried in vacuo to give a 1:1 mixture of 3- (1,3- dioxoindan-2-yl)benzoic acid, (M+H)+” 267, and 2- [2- (3- carboxyphenyl) acetyl] benzoic acid, (M+H)+‘ 285, (69.32 g) , which was used without further purification.
The mixture obtained in the previous step (52.8 g) was added to a solution of triethylamine (37.55 g, 0.372 mol) in industrial methylated spirit (500 ml) and the resulting cloudy solution was filtered through a pad of filter-aid to give a clear solution. Hydrazine monohydrate (9.3 g, 0.186 mol) was added in one portion at ambient temperature, the stirred mixture was heated under reflux for 1 hour, then it was concentrated in vacuo to approximately 250 ml and added to a solution of sodium acetate (41 g, 0.5 mol) in water (500 ml) . The mixture was brought to pH 7 by the dropwise addition of concentrated hydrochloric acid, then it was stirred at ambient temperature for 3 hours. The resulting solid was collected by filtration, washed with water (50 ml) and dried in va cuo to give a white solid (15.62 g) . The combined filtrate and washings were acidified to pH 6 by the addition of hydrochloric acid, then the mixture was stirred at ambient temperature for 3 hours. The resulting solid was collected by filtration, washed with water (50 ml) and dried in va cuo to give a second crop of off-white solid (17.57 g) . The combined filtrate and washings from the second crop were readjusted to pH 6 and treated as before to give a third crop of pale orange solid (6.66 g) . The three crops were combined to give essentially pure 3- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (A), (M+H)+‘ 281, δH 4.4 (2H, s), 7.2-7.4 (IH, m) , 7.5-7.6 (IH, ) , 7.7-8.0 (5H, m) , 8.1- 8.2 (IH, m) , 12.6 (IH, s)
b . 2-Fluoro-5- (4-oxo-3 , 4-dihydro-phthalazin -l -ylmethyl) benzoi c a cid (B)

Dimethyl phosphite (22.0 g, 0.2 mol) was added drop-wise to a solution of sodium methoxide (43.0 g) in methanol (100 ml) at 0°C. 2- Carboxybenzaldehyde (21.0 g, 0.1 mol) was then added portion-wise to the reaction mixture as a slurry in methanol (40 ml), with the temperature kept below 5°C. The resulting pale yellow solution was warmed to 20°C over 1 hour. Methanesulphonic acid (21.2 g, 0.22 mol) was added to the reaction drop-wise and the resulting white suspension was evaporated in va cuo . The white residue was quenched with water and extracted into chloroform (3 x 100 ml) . The combined organic extracts were washed with water (2 x 100 ml) , dried over MgS04, and evaporated in va cuo to yield (3-oxo-l, 3-dihydro-isobenzofuran-l-yl) phosphonic acid dimethyl ester as a white solid (32.0 g, 95 %, 95 % purity) . This was then used without further purification in the next stage.
To a mixture of (3-oxo-l, 3-dihydro-isobenzofuran-l-yl) phosphonic acid dimethyl ester (35.0 g, 0.14 mol) in tetrahydrofuran (200 ml) and 2- fluoro-5-formylbenzonitrile (20.9 g, 0.14 mol) in tetrahydrofuran (130 ml) was added triethylamine (14 ml, 0.14 mol) drop-wise over 25 min, with the temperature kept below 15°C. The reaction mixture was warmed slowly to 20°C over 1 hour and concentrated in vacuo . The white residue was slurried in water (250 ml) for 30 minutes, filtered, washed with water, hexane and ether, and dried to yield 2-fluoro-5- (3- oxo-3H-isobenzofuran-l-ylidenemethyl) benzonitrile as a 50:50 mixture of E and Z isomers (37.2 g, 96 %); m/z [M+l]+ 266 (98 % purity) To a suspension of 2-fluoro-5- (3-oxo-3H-isobenzofuran-l- ylidenemethyl) benzonitrile in water (200 ml) was added aqueous sodium hydroxide (26.1 g in 50 ml water) solution and the reaction mixture was heated under nitrogen to 90 °C for 30 minutes. The reaction mixture was partially cooled to 70°C, and hydrazine hydrate (100 ml) was added and stirred for 18 hours at 70°C. The reaction was cooled to room temperature and acidified with 2M HC1 to pH 4. The mixture was stirred for 10 min and filtered. The resulting solid was washed with water, hexane, ether, ethyl acetate and dried to yield 2-fluoro-5- (4-oxo-3, 4- dihydrophthalazin-l-ylmethyl)benzoic acid as a pale pink powder (30.0 g, 77 %) . m/z [M+l]+ 299 (96 % purity), δH 4.4 (2H, s) , 7.2-7.3 (IH, m) , 7.5-7.6 (IH, m) , 7.8-8.0 (4H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s).
c . 1 – [3- (4-Oxo-S , 4-dihydrophthalazin-l -ylmethyl) benzoyl]piperidine-4- carboxylic a cid (C)
 undesried????????
undesried????????(A) (C)
3- (4-Oxo-3, 4-dihydrophthalazin-l-ylmethyl)benzoic acid (A) (7.0 g, 0.25 mol), ethyl isonipecotate (5 ml, 0.32 mol), 2- (lH-benzotriazol-1-yl) – 1, 1, 3, 3-tetramethyluronium hexafluorophosphate (HBTU) (12.3 g, 0.32 mol) and N, N, -diisopropylethylamine (10.0 ml, 0.55 mol) were added to dimethylacetamide (40 ml) and stirred for 18 h. Water (100 ml) was added to the reaction mixture and the product was extracted into dichloromethane (4 x 50 ml) . The combined organic layers were washed with water (3 x 100 ml), dried over MgS0, filtered and evaporated in va cuo to yield an oil. To a solution of the oil in tetrahydrofuran (100 ml) was added 10 % aqueous sodium hydroxide solution (20 ml) and the reaction was stirred for 18 hours. The reaction was concentrated, washed with ethyl acetate (2 x 30 ml) and acidified with 2M HCl to pH 2. The aqueous layer was extracted with dichloromethane (2 x 100 ml), then the extracts were dried over MgS04, filtered and evaporated to yield 1- [3- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl)benzoyl]piperidine- 4-carboxylic acid (C) as a yellow solid (7.0 g, 65 %), m/z [M+l]+ 392
(96 % purity), δH 1.3-1.8 (5H, m) , 2.8-3.1 (4H, m) , .4 (2H, s), 7.2- 7.3 (IH, m) , 7.3-7.4 (IH, ) , 7.7-8.0 (5H, m) , 8.2-E 3 (IH, m) , 12.6 (IH, s) .
d . 1 – [2-Fluoro-5- (4 -oxo-3 , 4-dihydrophthala zin-l – ylmethyl) benzoyl]piperidine-4~carboxylic a cid (D)

(B) (D)
2-Fluoro-5- ( -oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (B) (3.1 g, 0.14 mol), ethyl isonipecotate (1.7 ml, 0.11 mol), 2-(lH- benzotriazol-1-yl) -1,1,3, 3-tetramethyluronium hexafluorophosphate (HBTU) (5.1 g, 0.13 mol) and N,N, -diisopropylethylamine (10.0 ml, 0.55 mol) were added to dimethylacetamide (15 ml) and stirred for 18 hours. Water (100 ml) was added to the reaction mixture and the product was extracted into dichloromethane (4 x 50 ml) . The combined organic layers were, filtered, washed with water (3 x 100 ml), dried over MgS04, filtered and evaporated in vacuo to yield an orange oil. The oil was purified by flash chromatography (ethyl acetate) to yield l-[2- fluoro-5- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoyl] piperidine-4- carboxylic acid as the methyl ester (1.5 g, 33 %, 96 % purity) . To a solution of the methyl ester in tetrahydrofuran: water (2:1, 40 ml) was added sodium hydroxide (0.3 g, 0.075 mol) and the reaction was stirred for 18 h. The reaction was concentrated, washed with ethyl acetate (2 x 20 ml) and acidified with 2M HC1 to pH 2. The aqueous layer was extracted with dichloromethane (2 x 20 ml) , and the combined extracts were dried over MgS04 and evaporated to yield 1- [3- ( 4-oxo-3, 4- dihydrophthalazin-1-ylmethyl) benzoyl] piperidine- -carboxylic acid (D) as a yellow solid (0.6 g, 65 %), m/z [M+l]+ 392 (96 % purity) Example 1 – Synthesis of Key Compounds
a. Synthesis of 4- [3- (piperazine-1-carfoonyl)benzyl] -2H-phthalasin-l- one (1)
 undesired????????
undesired????????(A) (1)
3- (4-0xo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (A) (5.0g, 0.17mol), tert-butyl 1-piperazinecarboxylate (3.9 g, 0.21 mol), 2-(lH- benzotriazol-1-yl) -1,1,3, 3-tetramethyluronium hexafluorophosphate (HBTU) (8.6 g, 0.22 mol) and N, , -diisopropylethylamine (6.7 ml, 0.38 mol) were added to dimethylacetamide (40 ml) and stirred for 18 hours. Water (100 ml) was added and the reaction mixture was heated to 100°C for 1 hour. The suspension was cooled to room temperature, filtered and dried to yield a white solid. The solid was dissolved in a solution of 6M HC1 and ethanol (2:1, 50 ml) and stirred for 1 hour. The reaction was concentrated, basified with ammonia to pH 9, and the product was extracted into dichloromethane (2 x 50 ml). The combined organic layers were washed with water (2 x 50 ml), dried over MgS04, and evaporated in va cuo to yield 4- [3- (piperazine-1-carbonyl) benzyl] – 2H-phthalazin-l-one (1) as a yellow crystalline solid (4.0 g, 77 %); m/z [M+l]+ 349 (97 % purity), δH 2.6-3.8 (8H, ) , 4.4 (2H, s), 7.2-7.5 (4H, m) , 7.7-8.0 (3H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s)
b . Synthesis of 4 – [4-Fluoro-3- (piperazine-1 -carbonyl) benzyl ] -2H- phthala zin ~l -one (2)
 desired……
desired……(β) (2)
The synthesis was carried out according to the method described in (a) above using 2-fluoro-5- (4-oxo-3, -dihydrophthalazin-l-ylmethyl) benzoic acid (B) to yield 4- [4-fluoro-3- (piperazine-1-carbonyl) benzyl] -2H- phthalazin-1-one (2) as a white crystalline solid (4.8 g, 76 %); m/z [M+l]+ 367 (97 % purity), δH 2.6-3.8 (8H, m) , 4.4 (2H, s), 7.2-7.5 (3H, m) , 7.7-8.0 (3H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s) .
…………………………..
US 8183369
http://www.google.co.in/patents/US8183369
4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (compound A) disclosed in WO 2004/080976:

is of particular interest.
A crystalline form of compound A (Form A) is disclosed in co-pending applications, which claim priority from U.S. 60/829,694, filed 17 Oct. 2006, entitled “Phthalazinone Derivative”, including U.S. Ser. No. 11/873,671 and WO 2008/047082.
Form A

References(a) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (Compound A)
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(15.23 g, 51.07 mmol) was suspended with stirring under nitrogen in acetonitrile (96 ml). Diisopropylethylamine (19.6 ml, 112.3 mmol) was added followed by 1-cyclopropylcarbonylpiperazine (I)(9.45 g, 61.28 mmol) and acetonitrile (1 ml). The reaction mixture was cooled to 18° C. 0-Benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (25.18 g, 66.39 mmol) was added over 30 minutes and the reaction mixture was stirred for 2 hours at room temperature. The reaction mixture was cooled to 3° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (20 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (20.21 g).
Mass Spectrum: MH+ 435
1H NMR (400 MHz, DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
………………………..
http://www.google.co.in/patents/US8247416
4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (compound A) disclosed in WO 2004/080976:

is of particular interest.
In WO 2004/080976, compound A was synthesised as one of a number of library compounds from 4-[4-fluoro-3-(piperazine-1-carbonyl)-benzyl]-2H-phthalazin-1-one (compound B):

by the addition of cyclopropanecarbonyl chloride:

to a solution of (B) in dichloromethane, followed by Hünig’s base (N,N-diisopropylethyl amine). This reaction is carried out with stirring at room temperature for 16 hours, and the resulting compound being purified by preparative HPLC.
The piperazine derivative (B) was prepared by deprotecting 4-[2-fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoyl]-piperazine-1-carboxylic acid tert-butyl ester (compound C):

by the use of 6M HCl and ethanol for 1 hour, followed by basification with ammonia to pH 9, and extraction into dichloromethane.
The Boc-protected piperazine derivative (C) was prepared from 2-fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoic acid (compound D):

by the addition of piperazine-1-carboxylic acid tert-butyl ester:

2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N,N,-diisopropylethylamine in dimethylacetamide, followed by stirring for 18 hours.
In WO 2004/080976, the following route to compound D is disclosed:

The method of synthesising compound D may further comprise the step of:
(c) synthesising 2-fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzonitrile (ED):

from compound E by reaction with hydrazine hydrate; and
(d) synthesising compound D from compound ED by reaction with sodium hydroxide.
Step (c) may be achieved by using between 1.1 and 1.3 equivalents of hydrazine hydrate in tetrahydrofuran followed by neutralisation of the excess hydrazine hydrate using acetic acid.
A sixth aspect of the present invention provides the compound ED:

and its use in the synthesis of compound D.
EXAMPLES
Example 1Synthesis of Compound A

Starting material (D) was synthesised by the method disclosed in WO 2004/080976
Methods
Preparative HPLC
Samples were purified with a Waters mass-directed purification system utilising a Waters 600 LC pump, Waters Xterra C18 column (5 μm 19 mm×50 mm) and Micromass ZQ mass spectrometer, operating in positive ion electrospray ionisation mode. Mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile) were used in a gradient; 5% B to 100% over 7 min, held for 3 min, at a flow rate of 20 ml/min.
Analytical HPLC-MS
Analytical HPLC was carried out with a Spectra System P4000 pump and Jones Genesis C18 column (4 μm, 50 mm×4.6 mm). Mobile phases A (0.1% formic acid in water) and B (acetonitrile) were used in a gradient of 5% B for 1 min rising to 98% B after 5 min, held for 3 min at a flow rate of 2 ml/min. Detection was by a TSP UV 6000LP detector at 254 nm UV and range 210-600 nm PDA. The Mass spectrometer was a Finnigan LCQ operating in positive ion electrospray mode.
(a) 4-[2-Fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoyl]-piperazine-1-carboxylic acid tert-butyl ester (C)
To a stirred solution of the starting material D (850 g) in dimethylacetamide (DMA) (3561 ml) at room temperature under nitrogen was added HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) (1402 g) in one portion. Hünig’s base (iPr2NEt, 1096 ml) was then added with the temperature kept between 15 to 25° C. followed by a solution of 1-Boc-piperazine (637 g) in DMA (1428 ml) with the temperature kept between 15 to 25° C.
The solution was stirred at room temperature for 2 hours and sampled for completion (HPLC). Upon completion the solution was added to vigorously stirred water (17085 ml) with the temperature kept between 15 to 25° C. and the solid filtered off, washing with water (2×7131 ml), hexane (2×7131 ml) and methyl tert-butyl ether (MTBE) (2×3561 ml). The solid was then dried overnight and then sampled for water content and chemical purity.
This reaction was then repeated, see table:
| Purity | Water Content | |||
| Batch | Yield (g) | (HPLC Area %) | (K.F.) | Corrected yield |
| 1 | 1571.3 | 86.80 | 24.3 | 1032.5 g (78%) |
| 2 | 2781.6 | 85.00 | 40.3 | 1411.5 g (106%) |
| a. Greater than 100% yield attributed to non-representative sampling | ||||
(b) 4-[4-Fluoro-3-(piperazine-1-carbonyl)-benzyl]-2H-phthalazin-1-one (B)
To a stirred solution of industrial methylated spirits (IMS) (2200 ml) and concentrated HCl (4400 ml) was added compound C (2780.2 g) in portions at room temperature under nitrogen, the foaming was controlled by the addition rate. The solution was then stirred at 15 to 25° C. for 30 minutes and sampled for completion (HPLC).
Upon completion the solution was evaporated to remove any IMS and the aqueous extracted with CH2Cl2 (2×3500 ml) before the pH was adjusted to >8 using concentrated ammonia. The resultant slurry was then diluted with water (10000 ml) and extracted with CH2Cl2 (4×3500 ml), washed with water (2×2000 ml), dried over MgSO4 (250 g) and evaporated. The crude product was then slurried in CH2Cl2 (3500 ml) and added to MTBE (5000 ml). The resultant suspension was filtered and dried at 50° C. overnight yielding 611.0 g (58.5% yield) of material with a purity of 94.12%
(c) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (A)
To a stirred suspension of compound B (1290 g) in CH2Cl2 (15480 ml) under nitrogen was added a pre-mixed solution of triethylamine (470 ml) and cyclopropane carbonyl chloride (306 ml) in CH2Cl2 (1290 ml) dropwise with the temperature kept below 20° C. The solution was then stirred at 10-15° C. for 15 minutes and sampled for completion. The reaction mixture was found to contain only 1.18% of starting material B and so the reaction was deemed complete and the batch was then worked-up.
The reaction mixture was washed with water (7595 ml), 5% citric acid solution (7595 ml), 5% sodium carbonate solution (7595 ml) and water (7595 ml). The organic layer was then dried over magnesium sulfate (500 g).
The CH2Cl2 containing product layer was then isolated, filtered through Celite and charged to a 251 vessel. CH2Cl2 (8445 ml) was then distilled out at atmospheric pressure and ethanol (10000 ml) added. Distillation was then continued with every 4000 ml of distillate that was removed being replaced with ethanol (4000 ml) until the head temperature reached 73.7° C. The reaction volume was then reduced (to 7730 ml) by which time the head temperature had reached 78.9° C. and the solution was allowed to cool to 8° C. overnight. The solid was then filtered off, washed with ethanol (1290 ml) and dried at 70° C. overnight. Yield=1377.3 g (90%). HPLC purity (99.34% [area %]). Contained 4.93% ethanol and 0.45% CH2Cl2 by GC.
(d) Water Treatment of Compound A
A suspension of compound A (1377.0 g), as produced by the method of Example 1, in water (13770 ml) was heated to reflux for 4 hours, cooled to room temperature and filtered. The solid was washed with water (2754 ml) and dried at 70° C. overnight. Yield=1274.8 g (92.6%). HPLC purity (99.49% [area %]). Contained 0.01% ethanol and 0.01% CH2Cl2 by GC.
1H NMR spectrum of compound A (DMSO-d6) following the water treatment is shown in FIG. 1.

The powder XRD pattern of Compound A following the water treatment is shown in FIG. 2, which shows the compound is as Form A.
Example 2
Alternative Synthesis of Compound A Using 1-(cyclopropylcarbonyl) piperazine

Methods (also for Examples 3 & 4)
NMR
1H NMR spectra were recorded using Bruker DPX 400 spectrometer at 400 MHz. Chemical shifts were reported in parts per million (ppm) on the δ scale relative to tetramethylsilane internal standard. Unless stated otherwise all samples were dissolved in DMSO-d6.
Mass Spectra
Mass spectra were recorded on an Agilent XCT ion trap mass spectrometer using tandem mass spectrometry (MS/MS) for structural confirmation. The instrument was operated in a positive ion elctrospray mode.
(a) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (Compound A)
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(15.23 g, 51.07 mmol) was suspended with stirring under nitrogen in acetonitrile (96 ml). Diisopropylethylamine (19.6 ml, 112.3 mmol) was added followed by 1-cyclopropylcarbonylpiperazine (1)(9.45 g, 61.28 mmol) and acetonitrile (1 ml). The reaction mixture was cooled to 18° C. O-Benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (25.18 g, 66.39 mmol) was added over 30 minutes and the reaction mixture was stirred for 2 hours at room temperature. The reaction mixture was cooled to 3° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (20 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (20.21 g).
Mass Spectrum: MH+435
1H NMR (400 MHz. DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
Example 3Alternative Synthesis of Compound A Using 1-(cyclopropylcarbonyl) piperazine HCl salt

(a) 1-(Cyclopropylcarbonyl)piperazine HCl salt (I′)
Acetic acid (700 ml) was treated with piperazine (50.00 g, 0.581 mol) portionwise over 15 minutes with stirring under nitrogen The reaction mixture was warmed to 40° C. and maintained at this temperature until a complete solution was obtained. Cyclopropanecarbonyl chloride 59.2 ml, 0.638 mol) was added over 15 minutes. The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered and the filtrate distilled under reduced pressure until ˜430 ml of distillates had been collected. Toluene (550 ml) was charged to the reaction mixture and reduced pressure distillation continued until a further 400 ml of distillates were collected. A further charge of toluene (550 ml) was added and reduced pressure distillation continued until 350 ml of distillates were collected. The resulting slurry was diluted with toluene (200 ml) and stirred overnight. Further toluene (500 ml) was added in order to mobilise the slurry. The slurry was filtered, washed with toluene (100 ml) and dried in vacuo at 40° C. to give the title compound as an off white solid (86.78 g).
Mass Spectrum: MH+155
1H NMR (400 MHz. D2O) δ: 0.92 (m, 4H), 1.98 (m, 1H), 3.29 (m, 2H), 3.38 (m, 2H), 3.84 (m, 2H), 4.08 (m, 2H).
(b) Compound A
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(0.95 g, 3.19 mmol) was suspended with stirring under nitrogen in acetonitrile (4 ml). 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (1.45 g, 3.83 mmol) was added followed by 1-cyclopropylcarbonylpiperazine HCl salt (I′)(0.73 g, 3.83 mmol). Diisopropylethylamine (1.39 ml, 7.98 mmol) was added over 3 minutes and the reaction mixture was stirred for overnight at room temperature. The reaction mixture was cooled to 5° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (2 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (0.93 g).
- “Olaparib, a PARP Inhibitor”. Health and Life.
- “AZ updates on olaparib and TC5214″. 20 December 2011.
- http://uk.reuters.com/article/2013/09/04/astrazeneca-cancer-idUKL6N0H00KN20130904
- http://www.clinicaltrials.gov/ct2/show/NCT01844986
- New cancer drug ‘shows promise’ BBC News 24 June 2009
- Olaparib for the treatment of ovarian cancer.
- Vasiliou S, Castaner R, Bolos J. Olaparib. Drugs of the Future. 2009; 34(2): 101.
- Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S, Javaid H, Kerrigan F, Knights C, Lau A, Loh VM, Matthews IT, Moore S, O’Connor MJ, Smith GC, Martin NM (October 2008). “4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1″. Journal of Medicinal Chemistry 51 (20): 6581–91. doi:10.1021/jm8001263. PMID 18800822.
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O’Connor MJ, Martin NM, Borst P, Jonkers J (November 2008). “High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs”. Proceedings of the National Academy of Sciences of the United States of America 105 (44): 17079–84. doi:10.1073/pnas.0806092105. PMC 2579381. PMID 18971340.
- Hay T, Matthews JR, Pietzka L, Lau A, Cranston A, Nygren AO, Douglas-Jones A, Smith GC, Martin NM, O’Connor M, Clarke AR (May 2009). “Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin”. Cancer Research 69 (9): 3850–5. doi:10.1158/0008-5472.CAN-08-2388. PMID 19383921.
- http://www.ncri.org.uk/ncriconference/archive/2007/abstracts/pdf/LB57.pdf “A Phase I trial of AZD2281 (KU-0059436), a PARP inhibitor with single agent anticancer activity in patients with BRCA deficient tumours, particularly ovarian cancer”
- Fong PC, Boss DS, Yap TA, et al. (July 2009). “Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers”. N. Engl. J. Med. 361 (2): 123–34.doi:10.1056/NEJMoa0900212. PMID 19553641.
- http://www.cancercompass.com/cancer-news/1,15869,00.htm “Phase II Trials Investigating Oral PARP Inhibitor, Olaparib, In BRCA-Deficient Advanced Breast And Ovarian Cancer” June 2009
- http://clinicaltrials.gov/ct2/show/NCT00912743 Efficacy and Safety of Olaparib in Pretreated Patients With Measurable Colorectal Cancer, Stratified by Microsatellite Instability (MSI) Status
- “Olaparib Looks Promising in Treatment of Non-BRCA Ovarian Cancer”. 26 Aug 2011.
| Patent | Submitted | Granted |
|---|---|---|
| Phthalazinone Derivatives [US2012010204] | 2012-01-12 | |
| PARP1 TARGETED THERAPY [US2012035244] | 2012-02-09 | |
| Phthalazinone derivatives [US7449464] | 2005-03-17 | 2008-11-11 |
| 4- [3- (4-CYCLOPROPANECARBONYL-PIPERAZINE-I-CARBONYL) -4 -FLUORO-BENZYL] -2H-PHTHALAZ IN-1-ONE [US8183369] | 2010-11-11 | 2012-05-22 |
| PHTHALAZINONE DERIVATIVES [US7692006] | 2008-06-19 | 2010-04-06 |
| PHTHALAZINONE DERIVATIVES [US7981889] | 2008-08-21 | 2011-07-19 |
| PHARMACEUTICAL FORMULATION 514 [US2010098763] | 2010-04-22 | |
| PHTHALAZINONE DERIVATIVE [US8247416] | 2009-10-29 | 2012-08-21 |
| WO2002036576A1 * | 25 Oct 2001 | 10 May 2002 | Kudos Pharm Ltd | Phthalazinone derivatives |
| WO2002090334A1 * | 30 Apr 2002 | 14 Nov 2002 | Kudos Pharm Ltd | Isoquinolinone derivatives as parp inhibitors |
| WO2003093261A1 * | 29 Apr 2003 | 13 Nov 2003 | Kudos Pharm Ltd | Phthalazinone derivatives |
extras…………..
|
|
| Systematic (IUPAC) name | |
|---|---|
| 4-[(3-[(4-cyclopropylcarbonyl)piperazin-4-yl]carbonyl) -4-fluorophenyl]methyl(2H)phthalazin-1-one | |
| Clinical data | |
| Trade names | Lynparza |
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 763113-22-0  |
| ATC code | None |
| PubChem | CID 23725625 |
| ChemSpider | 23343272  |
| UNII | WOH1JD9AR8 |
| ChEMBL | CHEMBL521686 |
| Chemical data | |
| Formula | C24H23FN4O3 |
| Mol. mass | 435.08 g/mol |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
REFERENCES
nmr
 CAS NO. 763113-22-0, olaparib H-NMR spectral analysis |
 CAS NO. 763113-22-0, olaparib C-NMR spectral analysis |
Filed under: 0rphan drug status, cancer Tagged: ASTRAZENECA, AZD-2281, オラパリブ, CANCER, Lynparza, olaparib, Orphan Drug, ovarian cancer, The European Commission, 奥拉帕尼







