![Bortezomib.svg]()
BORTEZOMIB
A proteasome inhibitor.
EMA:Link,
US FDA:link
[(1R)-3-methyl-1-({(2S)-3-phenyl-2-[(pyrazin-2-ylcarbonyl)amino]propanoyl}amino)butyl]boronic acid
Λ/-(pyrazin-2-yl)carbonyl-L-phenylalanine-L-leucine boronic acid
179324-69-7 CAS
- Bortezomib
- HSDB 7666
- LDP 341
- LDP-341
- MG 341
- MLN-341
- NSC 681239
- PS 341
- PS 341 (pharmaceutical)
- PS-341
- UNII-69G8BD63PP
- Velcade
For treatment of multiple myeloma in patients who have not been successfully treated with at least two previous therapies.
A dipeptide boronic acid analogue with antineoplastic activity. Bortezomib reversibly inhibits the 26S proteasome, a large protease complex that degrades ubiquinated proteins. By blocking the targeted proteolysis normally performed by the proteasome, bortezomib disrupts various cell signaling pathways, leading to cell cycle arrest, apoptosis, and inhibition of angiogenesis. Specifically, the agent inhibits nuclear factor (NF)-kappaB, a protein that is constitutively activated in some cancers, thereby interfering with NF-kappaB-mediated cell survival, tumor growth, and angiogenesis. In vivo, bortezomib delays tumor growth and enhances the cytotoxic effects of radiation and chemotherapy. (NCI Thesaurus)
Bortezomib (originally PS-341 and marketed as Velcade by Millennium Pharmaceuticals) is the first therapeutic proteasome inhibitor to be tested in humans. It is approved in the U.S. for treating relapsed multiple myeloma and mantle cell lymphoma. In multiple myeloma, complete clinical responses have been obtained in patients with otherwise refractory or rapidly advancing disease.
![]() bortezomib
bortezomib
![]()
Bortezomib (BAN, INN and USAN. Originally codenamed PS-341; marketed as Velcade by Millennium Pharmaceuticals and Cytomib by Venus Remedies) is the first therapeutic proteasome inhibitor to be tested in humans. It is approved in the U.S. for treating relapsed multiple myeloma[1] andmantle cell lymphoma. In multiple myeloma, complete clinical responses have been obtained in patients with otherwise refractory or rapidly advancing disease.
Bortezomib was originally synthesized in 1995 (MG-341) at a company called Myogenics, which soon changed its name to ProScript. After promising preclinical results, the drug (PS-341) was tested in a small Phase I clinical trial on patients with multiple myeloma cancer. ProScript ran out of money and was bought by Leukosite in May 1999. Leukosite in turn was bought by Millennium Pharmaceuticals in October 1999. At this point in time, the project had low priority amongst other projects at the company. This changed significantly when one of the first volunteers to receive the drug in the clinical trial achieved a complete response and were still alive four years later. At the time this was a remarkable result. Later clinical experimentation indicates the possibility of a complete response in 15% of patients in a similar condition, when treated with bortezomib.
In May 2003, seven years after the initial synthesis, bortezomib (Velcade) was approved in the United States by the Food and Drug Administration(FDA) for use in multiple myeloma, based on the results from the SUMMIT Phase II trial.[2]
Another commercially available bortezomib product – Bortenat (Natco Pharma, India), reportedly contains substantially more active entity than declared, potentially and even more resulting in increase toxicity. Moreover, Bortenat has some other chemical and formulation deviations from the registered ethic product Velcade (Millennium Pharmaceuticals and Janssen-Cilag), with unclear clinical impact.[3]
![]()
Bortezomib bound to the core particle in a yeast proteasome. The bortezomib molecule is in the center colored by atom type (boron = pink, carbon = cyan, nitrogen = blue, oxygen = red), surrounded by the local protein surface. The blue patch is catalyticthreonine residue whose activity is blocked by the presence of bortezomib.
VELCADE® (bortezomib) for Injection is an antineoplastic agent available for intravenous injection or subcutaneous use. Each single use vial contains 3.5 mg of bortezomib as a sterile lyophilized powder. Inactive ingredient: 35 mg mannitol, USP.
Bortezomib is a modified dipeptidyl boronic acid. The product is provided as a mannitol boronic ester which, in reconstituted form, consists of the mannitol ester in equilibrium with its hydrolysis product, the monomeric boronic acid. The drug substance exists in its cyclic anhydride form as a trimeric boroxine.
The chemical name for bortezomib, the monomeric boronic acid, is [(1R)-3-methyl-1-[[(2S)-1-oxo-3-phenyl-2[(pyrazinylcarbonyl) amino]propyl]amino]butyl] boronic acid.
Bortezomib has the following chemical structure:
The molecular weight is 384.24. The molecular formula is C19H25BN4O4. The solubility of bortezomib, as the monomeric boronic acid, in water is 3.3 to 3.8 mg/mL in a pH range of 2 to 6.5.
Structure
The drug is an N-protected dipeptide and can be written as Pyz-Phe-boroLeu, which stands for pyrazinoic acid,phenylalanine and Leucine with a boronic acid instead of a carboxylic acid. Peptides are written N-terminus to C-terminus, and this convention is used here even though the “C-terminus” is a boronic acid instead of a carboxylic acid.
-
Boronic acid and ester compounds display a variety of pharmaceutically useful biological activities.
Shenvi et al., U.S. Pat. No. 4,499,082 (1985 ), discloses that peptide boronic acids are inhibitors of certain proteolytic enzymes.
Kettner and Shenvi, U.S. Pat. No. 5,187,157 (1993 ),
U.S. Pat. No. 5,242,904 (1993 ), and
U.S. Pat. No. 5,250,720 (1993 ), describe a class of peptide boronic acids that inhibit trypsin-like proteases.
Kleeman et al., U.S. Pat. No. 5,169,841 (1992 ), discloses
N-terminally modified peptide boronic acids that inhibit the action of renin.
Kinder et al., U.S. Pat. No. 5,106,948 (1992 ), discloses that certain tripeptide boronic acid compounds inhibit the growth of cancer cells.
-
More recently, boronic acid and ester compounds have displayed particular promise as inhibitors of the proteasome, a multicatalytic protease responsible for the majority of intracellular protein turnover.Ciechanover, Cell, 79: 13-21 (1994), discloses that the proteasome is the proteolytic component of the ubiquitin-proteasome pathway, in which proteins are targeted for degradation by conjugation to multiple molecules of ubiquitin. Ciechanover also discloses that the ubiquitin-proteasome pathway plays a key role in a variety of important physiological processes.
-
Adams et al., U.S. Patent No. 5,780,454 (1998 ),
U.S. Patent No. 6,066,730 (2000 ),
U.S. Patent No. 6,083,903 (2000 ),
U.S. Patent No. 6,297,217 (2001 ),
U.S. Patent No. 6,548,668 , and
U.S. Patent No. 6,617,317 (2003 ), hereby incorporated by reference in their entirety, describe peptide boronic ester and acid compounds useful as proteasome inhibitors. The references also describe the use of boronic ester and acid compounds to reduce the rate of muscle protein degradation, to reduce the activity of NF-κB in a cell, to reduce the rate of degradation of p53 protein in a cell, to inhibit cyclin degradation in a cell, to inhibit the growth of a cancer cell, to inhibit antigen presentation in a cell, to inhibit NF-κB dependent cell adhesion, and to inhibit HIV replication.
-
Albanell and Adams, Drugs of the Future 27: 1079-1092 (2002), discloses that one such peptide boronic acid proteasome inhibitor, bortezomib (N-2-pyrazinecarbonyl-L-phenylalanine-L-leucineboronic acid), shows significant antitumor activity in human tumor xenograft models and is undergoing clinical evaluation. Richardson et al., New Engl. J. Med., 348:2609 (2003), reports the results of a Phase 2 study of bortezomib, showing its effectiveness in treating relapsed and refractory multiple myeloma.
-
Studies of boronic acid protease inhibitors have been greatly advanced by the development of chemistry for the preparation of functionalized boronic acid compounds, particularly alpha-halo- and alpha-aminoboronic acids. Matteson and Majumdar, J. Am. Chem. Soc., 102:7590 (1980), discloses a method for preparing alpha-chloroboronic esters by homologation of boronic esters, and Matteson and Ray, J. Am. Chem. Soc., 102:7591 (1980), reports that chiral control of the homologation reaction can be achieved by the use of pinanediol boronic esters. The preparation of alpha-aminoboronic acid and ester compounds from the corresponding alpha-chloroboronic esters has also been reported (Matteson et al., J. Am. Chem. Soc., 103:5241 (1981);
Shenvi, U.S. Patent No. 4,537,773 (1985 )).
-
Matteson and Sadhu, U.S. Patent No. 4,525,309 (1985 ), describes an improved procedure for the homologation of boronic esters by rearrangement of the intermediate boron “ate” complex in the presence of a Lewis acid catalyst. The Lewis acid is reported to promote the rearrangement reaction and to minimize epimerization at the alpha-carbon atom. Rigorous exclusion of water and careful control of Lewis acid stoichiometry are required for optimum results, however. These features render the reaction difficult to perform successfully on a production scale, and limit the availability of pharmaceutically important boronic ester and acid compounds, such as bortezomib
The boron atom in bortezomib binds the catalytic site of the 26S proteasome[4] with high affinity and specificity. In normal cells, the proteasome regulates protein expression and function by degradation of ubiquitylated proteins, and also cleanses the cell of abnormal or misfolded proteins. Clinical and preclinical data support a role in maintaining the immortal phenotype of myeloma cells, and cell-culture and xenograft data support a similar function in solid tumor cancers. While multiple mechanisms are likely to be involved, proteasome inhibition may prevent degradation of pro-apoptotic factors, permitting activation of programmed cell death in neoplastic cells dependent upon suppression of pro-apoptotic pathways. Recently, it was found that bortezomib caused a rapid and dramatic change in the levels of intracellular peptides that are produced by the proteasome.[5] Some intracellular peptides have been shown to be biologically active, and so the effect of bortezomib on the levels of intracellular peptides may contribute to the biological and/or side effects of the drug.
![]() BORTEZOMIB
BORTEZOMIB
Bortezomib is rapidly cleared following intravenous administration.[6] Peak concentrations are reached at about 30 minutes. Drug levels can no longer be measured after an hour.Pharmacodynamics are measured by measuring proteasome inhibition in peripheral blood mononuclear cells. The much greater sensitivity of myeloma cell lines and mantle cell lines to proteasome inhibition compared with normal peripheral blood mononuclear cells and most other cancer cell lines is poorly understood.
Costs
UK
NICE recommended against Velcade in Oct 2006 due to its cost.[7]
The company proposed a cost reduction for multiple myeloma,[8] and this was taken up in the UK.[9]
![]()
Bortezomib is associated with peripheral neuropathy in 30% of patients; occasionally, it can be painful. This can be worse in patients with pre-existing neuropathy. In addition, myelosuppressioncausing neutropenia and thrombocytopenia can also occur and be dose-limiting. However, these side effects are usually mild relative to bone marrow transplantation and other treatment options for patients with advanced disease. Bortezomib is associated with a high rate of shingles,[10] although prophylactic acyclovir can reduce the risk of this.[11]
Gastro-intestinal (GI) effects and asthenia are the most common adverse events.[12]
Green tea extract epigallocatechin gallate (EGCG), which had been expected to have a synergistic effect, was found by Encouse B. Golden, et al. to reduce the effectiveness of bortezomib.[13][14][15][16]
Two open-label, phase II trials (SUMMIT and CREST) established the efficacy of bortezomib 1.3 mg/m2 (with or without dexamethasone) administered by intravenous bolus on days 1,4,8, and 11 of a 21-day cycle for a maximum of eight cycles in heavily pretreated patients with relapsed/refractory multiple myeloma.[17] The phase III APEX trial demonstrated the superiority of bortezomib 1.3 mg/m2 over a high-dose dexamethasone regimen (e.g. median TTP 6.2 vs 3.5 months, and 1-year survival 80% vs 66%).[17]
PATENTS
| Canada |
2203936 |
2005-04-12 |
EXPIRY2015-10-27 |
| United States |
6713446 |
2002-01-25 |
2022-01-25 |
| United States |
6083903 |
1994-10-28 |
2014-10-28 |
Raghavendracharyulu Venkata Palle, Rajasekhar Kadaboina, Veerendeer Murki, Amarendhar Manda, Nageshwar Gunda, Ramaseshagiri Rao Pulla, Mallesha Hanmanthu, Narasimha Naidu Mopidevi, Suresh Kumar Ramdoss, “BORTEZOMIB AND PROCESS FOR PRODUCING SAME.” U.S. Patent US20100226597, issued September 09, 2010.
![]()
INTRODUCTION
Bortezomib (PS-341, Velcade®; N-(pyrazin-2-yl)carbonyl-L-phenylalanine-L-leucine boronic acid; (1R)-3-Methyl-1-[(2S)-3-phenyl-2-[(pyrazinylcarbonyl)amino]propanoyl]amino]butyl]boronic acid; CAS Registry Number: 179324-69-7) is an N-acylated dipeptide analogue of phenylalanyl-leucine in which a boronic acid functional group replaces the C-terminal carboxylic acid. It is a white to almost white crystalline powder and when appropriately formulated for injection is an anti-neoplastic agent and is a therapeutic proteosome inhibitor. In the US this active pharmaceutical ingredient (API) is approved for the treatment of multiple myeloma and mantle cell lymphoma.
Bortezomib is composed of three moieties that are fused together by two amide bonds. Two of these three units can be thought of as analogues of amino acids (viz., an α-aminoboronic acid and a pyrazinecarboxylic acid) and the third unit is a naturally occurring amino acid (viz., L-phenylalanine). Bortezomib possesses two chiral centres but is a single stereoisomer. One chiral centre exists within the α-aminoboronic acid moiety and the other exists within the naturally occurring amino acid, L-phenylalanine, moiety. In the solid state under anhydrous conditions, bortezomib can exist as a trimeric anhydride (trimeric boroxine), herein referred to as bortezomib anhydride. In the presence of water this can be hydrolysed to its monomeric boronic acid form.
![Figure US20120289699A1-20121115-C00001]()
Amino boronic acids – amino acids wherein terminal carboxylic groups are replaced by boronic B(OH)2 groups – are important pharmacoisosters of amino acids in various therapeutically promising molecules, mainly for treatment of cancer. For instance, talabostat contains proline boronic acid, bortezomib contains leucine boronic acid. Bortezomib, chemically Λ/-(pyrazin-2-yl)carbonyl-L-phenylalanine-L-leucine boronic acid, is an important proteasome inhibitor and has been clinically approved for use in treating mantle cell lymphoma and multiple myeloma. Recently, many novel molecules containing amino boronic acids, especially leucine boronic acid, have been prepared and biologically tested as described in WO2009/006473 A2.
The synthesis of bortezomib and other amino boronic acid and ester compounds is disclosed in
EP0788360 B1 , international patent application WO2005/097809 A2, international patent application
WO2009/004350 A1 , and international patent application WO2009/036281 A2.
EP0788360 B1 describes a general process for preparation of amino boronic acid and ester compounds using (1 S, 2S, 3R, 5S)-pinanediol leucine boronate and an amino acid or its derivative as starting materials. As coupling agents 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide hydrochloride (EDC), benzotriazol-1-yloxytris (dimethylamino) phosphonium hexafluorophosphate (BOP reagent), or 0-(MH- benzotriazol-1-yl)-/V,/V,/V,/V-tetramethyluronium tetrafluoroborate (TBTU) were employed.
A synthetic process suitable for a large scale production of amino boronic acid and ester compounds is described in WO2005/097809 A2. The synthesis involves a boronate complex, which is contacted with a
Lewis acid under conditions that afford the boronic ester compounds.
WO2009/004350 A1 discloses a high yield synthesis of bortezomib and intermediates for the synthesis thereof. The procedure includes the use of a very high percentage of tetrahydrofuran in the halogenation of the starting compound (S)-pinanediol 2-m ethyl propane- 1 -boronate.
WO2009/036281 A2 describes processes for the preparation of substantially pure bortezomib and intermediates thereof. Processes for the preparation of crystalline forms of bortezomib as well as a storage system for bortezomib are also disclosed in said patent application.
In international patent application WO2005/097809 A2, in J. Biol. Chem. 1984, 259, 15106-15114 and in J. Am. Chem. Soc. 1981 , 103, 5241-5242 a route for the preparation of α-amino boronic esters, which is known to the person skilled in the art known as the Matteson’s synthetic route, is described. Homologation of boronic esters with (dichloromethyl)lithium to form α-chloro boronic esters has been shown to be efficient and result in good chiral selectivity if pinanediol was used as the chiral directing group. The use of the Lewis acid (ZnCI2) as a catalyst and chloride ion scavenger for the rearrangement of the borate intermediate improved the diastereomeric ratio in the α-chloro boronic ester product, α- Chloro boronic esters have been converted to silylated α-amino boronic esters by lithiumhexamethyldisilazane (LiHMDS), which have been desilylated and protonated in situ to the α-amino boronic esters.
An approach for the synthesis of diverse α-amino boronic esters by the highly diastereoselective copper- catalyzed addition of bis(pinacolato)diboron to N-tert-butane sulfinyl aldimines has been disclosed in the J. Am. Chem. Soc. 2008, 730, 6910-6911.
Transformation of 1 ,1-dihalogenoalkenes to corresponding alkynes and subsequent synthesis of 1- alkynylboranes have been described in Tetrahedron Letters 1972, 13, 3769-3772 and Tetrahedron Letters 1988, 29, 2631-2634.
J. Am. Chem. Soc. 1994, 116, 10302-10303 describes a process for preparing α-substituted 1- alkenyldioxaborolanes starting from 1-alkynyldioxaborolanes by hydrozirconation followed by substitution such as halogenation or carbonylation. The following α-substituted 1-alkenyldioxaborolanes are disclosed in this reference: (E)-2-(1-chloro-3,3-dimethylbut-1-enyl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane, (E)-2- (1-bromo-3,3-dimethylbut-1-enyl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane, (E)-2-(1-iodo-3,3-dimethylbut- 1-enyl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane, (E)-2,2,6,6-tetramethyl-4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)hept-4-en-3-one, (E)-4,4-dimethyl-1-phenyl-2-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)pent-2-en-1-one, (E)-2-(4,4-dimethylpent-2-en-2-yl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane zircono- cene and (E)-2-(hept-2-en-2-yl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane zirconocene.
Bortezomib has developed a firm Millennium Pharmaceuticals in 2004 [3]. The drug has proved such a success that in record time has been approved by the FDA for the treatment of multiple myeloma and lymphoma. As can be seen easily bortezomib is a derivative modified dipeptide Phe-Leu-B (OH) 2 . The final step of the synthesis is not requiring comment passage
……………………………………………………
WO 2005/097809
![]() SYNTHESIS/US20120289699
SYNTHESIS/US20120289699
The chiral centre of the α-aminoboronic acid moiety cannot, however, be derived from a chiral pool since α-aminoboronic acids are not known to be naturally occurring.
Instead, enantio-enriched α-aminoboronic acids in which the chiral centre is adjacent to the boron atom can be obtained by the use of chiral boron chemistry developed by Matteson, such as disclosed in U.S. Pat. No. 4,525,309 and a series of peer reviewed publications. Matteson’s chemistry when used for chiral applications utilises a boronic ester comprising a chiral diol auxiliary (such as 1S,2S,3R,5S)-(+)-2,3-pinanediol ((S)-(+)-pinanediol), for example) which upon reaction with the lithium salt (this salt can be prepared in situ or separately) of dichloromethane forms an α,α-dichloroboronate complex, which the boron ate functional group is chiral.
Due to induction provided by the chiral diol auxiliary, the boron ate complex undergoes a spontaneous and stereoselective internal rearrangement with displacement of one of the prochiral chloro substituents to generate an α-chloroboronic ester which possesses a newly generated chiral centre adjacent to the boron atom (See Scheme 1). This rearrangement of the boron ate complex is dramatically improved by catalysis with ZnCl2 (see J. Am. Chem. Soc., 1983, 105, 2077-2078).
α-Chloroboronic esters can be converted into the aforementioned requisite α-aminoboronic acids, preferably protected as boronic esters, possessing useful high chiral purity by reaction with LiHMDS followed by desilylation and optional salt formation of the amino group). Altogether, this reaction sequence provides a 1-carbon homologation of the original carbon backbone of the B-alkyl portion of the boronic ester in addition to a stereoselectively appended amino group. Most typically the chiral auxiliary demonstrated for this reaction sequence is homochiral pinanediol, such as the (+)-enantiomer referred to as (S)-(+)-pinanediol, or the (−)-enantiomer referred to as (R)-(−)-pinanediol.
![Figure US20120289699A1-20121115-C00002]()
One drawback with this stereoselective approach to α-aminoboronic acid synthesis in an industrial setting is the relatively high cost of the chiral diol auxiliary, pinanediol. Further, the use of the chiral diol imposes other synthetic restrictions, such as order of installation of the alkyl group to be homologated (i.e., the R group and dichloromethyl substituent) into the boron ate complex, and the relatively more difficult hydrolysis step required to remove stereochemically hindered diol groups afterwards. Despite this U.S. Pat. No. 7,714,159 B2, WO2009004350A1 and WO2009036281A2 disclose methods for the synthesis of bortezomib utilizing Matteson’s chemistry in conjunction with (S)-(+)-pinanediol as the chiral auxiliary.
Although a chiral auxiliary, such as (S)-(+)-pinanediol, is required for chiral induction in the homologation step in the Matteson reaction sequence, a chiral auxiliary itself is not inherently required for the Matteson homologation step to proceed, and achiral diols can also be used (Organometallics, 1983, 2, 1529-1535).
The inventors of the invention herein reasoned that the high cost of (S)-(+)-pinanediol could be circumvented in the synthesis of bortezomib by the use of a cheap, achiral diol to protect the boronic acid functional group. Since the use of an achiral diol auxiliary would not provide any stereochemical induction in the homologation step, a racemic product (that is, it would comprise equimolar amounts of each enantiomer) would be produced, which itself or a down stream synthetic derivative of it would require a classical resolution or other technique capable of separating the stereoisomers to be performed upon it.
Given that there was a need for a separation method that could separate the racemate, the inventors reasoned that one efficient approach would utilise the enantiopure API building block, L-phenylalanine as an intramolecular chiral resolving agent. L-phenylalanine or its derivatives could serve as a cost efficient in-process chiral resolving agent in this manner because i) it and its derivatives are cheap and are commercially available on large scales, and ii) it comprises part of the molecular structure of bortezomib itself. Therefore it was reasoned that its use would not be wasteful once the desired enantiomer of the racemate was separated because it would also be incorporated into the API itself.
Thus, a key characteristic of the invention herein useful for the synthesis of bortezomib is the use of a racemic diol α-aminoboronic ester salt, such as the pinacol derivative [5], as a key intermediate. This racemic key intermediate is derivatised by its reaction with L-phenylalanine to provide a mixture of diastereomers that are separated by crystallisation, or by chromatography, or by stereoselective hydrolysis.
The requisite racemic boronic esters, such as pinacol α-aminoboronic ester [5], are readily synthesized utilizing prior art chemistry disclosed by Matteson (e.g., see Pure & Appl. Chem., 1985, 57, 1741-1748), as exemplified in Scheme 2.
The racemic boronic esters, such as the pinacol α-aminoboronic ester [5], are then converted into mixtures of diastereomers [6] by reaction with a suitably protected L-phenylalanine derivative (See Scheme 3), such as N-BOC-L-phenylalanine. The protecting group of the L-phenylalanine moiety is then removed, such as by reacting the diastereomers [6] with an acid such as hydrochloric acid, to form a mixture of amine salt diastereomers [7] which is then subjected to conditions under which the desired diastereomer (R,S)-[7] is selectively isolated, such as by crystallisation, chromatography or stereoselective hydrolysis. The separated desired diastereomer (R,S)-[7] is then converted into bortezomib or bortezomib anhydride.
![Figure US20120289699A1-20121115-C00004]()
In this invention the need for the use of an expensive chiral auxiliary such as (S)-(+)-pinanediol to induce stereoselectivity in the Matteson homologation reaction sequence is circumvented by the use of the naturally occurring and relatively cheap amino acid L-phenylalanine in protected form. In addition to being 7-10 times cheaper than (S)-(+)-pinanediol, unlike (S)-(+)-pinanediol which is liberated from the penultimate API precursor at the end of the synthesis of bortezomib following the methods of the prior art, the amino acid, L-phenylalanine, comprises part of the final API molecular structure.
This invention differs from those disclosed in U.S. Pat. No. 7,714,159 B2, WO2009004350A1 and WO2009036281A2 which all rely on the use of the expensive chiral diol auxiliary (S)-(+)-pinanediol in conjunction with Matteson chemistry to obtain the requisite chirality.
EXAMPLES
For embodiment 1, as mentioned previously, the process has been demonstrated using pinacol as the boronate ester diol moiety and the hydrochloride salt of the diastereomeric mixture of [7].
Example 1 Synthesis of [7] Pinacol 1-chloro-3-methylbutane-1-boronate (rac-[3])
A mixture of THF (2 L) and DCM (55.3 g, 0.651 mol) was cooled to −100° C. n-BuLi (260.7 mL, 2.5 M in n-hexane, 0.651 mol) was added dropwise into the reaction mixture maintaining at −100° C. Pinacol 2-methylpropane-1-boronate ([2]; 100 g, 0.543 mol) was added into the reaction mixture. The resulting mixture was keep at −100° C. for one hour. A solution of ZnCl2 (136.3 g, 1.0 mol) in THF (500 mL) was added dropwise to the reaction over a period of 60 minutes. The resulting mixture was keep at −100° C. for one hour, the reaction mixture was warmed up to room temperature and keep at room temperature overnight. The reaction was diluted with MTBE (750 mL) and was washed twice with saturated NH4Cl (2 L each). The organic layer was dried overnight over MgSO4 before filtering and evaporating. n-Heptane (250 mL) was added into the mixture and was filtered and evaporated providing the product as an oil (119.5 g, 0.514 mol). 1H NMR (300 MHz, CDCl3) δ 3.48 (dd, J=9.8, 6.1 Hz, 1H), 1.93-1.71 (m, 2H), 1.61 (td, J=8.1, 4.0 Hz, 1H), 1.33-1.24 (m, 12H), 0.95-0.87 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 84.5, 42.8, 25.8, 24.8, 23.1, 21.5.
Pinacol 1-bis-(trimethylsilyl)-amino-3-butane-1-boronate (rac-[4])
A solution of LiHMDS (44.61 g, 0.267 mol in 217 mL THF) in THF (750 mL) was cooled to −75° C. and pinacol 1-chloro-3-methylbutane-1-boronate (rac-[3]; 77.5 g, 0.333 mol) in THF (462 mL) was added. The resulting mixture was keep at −75° C. for 1 hour. The reaction mixture was warmed up to room temperature and kept at room temperature overnight. The mixture was evaporated to provide the product as an oil (73 g, 0.204 mol). 1H NMR (300 MHz, CDCl3) δ 2.58 (t, J=7.7 Hz, 1H), 1.75 (tq, J=13.1, 6.5 Hz, 1H), 1.66-1.44 (m, 1H), 1.34-1.27 (m, 1H), 1.22 (s, 12H), 0.90-0.84 (m, 6H), 0.12-0.09 (m, 18H).
Pinacol-1-ammonium chloride-3-methylbutane-1-boronate (rac-[5])
A solution of pinacol 1-bis-(trimethylsilyl)-amino-3-butane-1-boronate (rac-[4]; 264.9 g, 0.741 mol) in n-heptane (4 L) and diethyl ether (1.6 L) was cooled to −35° C. HCl gas was bubbled through the mixture for 4 hours, and the resulting mixture was stirred at room temperature overnight and was then filtered. The filter cake was dissolved in DCM (1 L) and was stirred at room temperature for 2.5 hours, filtered and evaporated. The residue was diluted with EtOAc (713 mL) to form a slurry that was stirred for 1 hour and then filtered. The solid was dried under vacuum at 35° C. to provide the product as white crystals (123.9 g, 0.496 mol). 1H NMR (300 MHz, d6-DMSO) δ 7.75 (s, 3H), 2.70 (d, J=5.5 Hz, 1H), 1.68 (dt, J=13.5, 6.8 Hz, 1H), 1.44 (t, J=7.3 Hz, 2H), 1.24 (s, 12H), 0.86 (d, J=6.5 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 85.0, 38.6, 35.9, 25.1, 24.8, 22. 5; ESI-MS (positive) (m/z): 213, 170, 156, 128, 100, 88, 74.
Pinacol N-BOC-L-phenylalanine-D,L-leucine boronate ((R,S)-/(S,S)-[6])
To a cooled (about 0° C.) solution of BOC-L-phenylalanine (60.6 g, 0.228 mol) in DMF (670 mL) was added DIPEA (62.1 g, 0.480 mol), HATU (96.0 g, 0.252 mol) and a DMF (290 mL) solution of rac-[5] (pinacol-1-ammonium chloride-3-methylbutane-1-boronate) (60 g, 0.240 mol). The mixture was warmed to room temperature and stirred at this temperature overnight. Ethyl acetate (1 L) and a saturated aqueous solution of sodium of chloride (700 mL) were added into the reaction mixture. After mixing, the organic layer was separated and washed with a saturated aqueous solution of sodium of chloride (750 mL), then with an aqueous 0.1 N solution of KHSO4 (800 mL) and finally with an saturated aqueous solution of NaHCO3 (800 mL). The organic layer was dried over MgSO4 and concentrated at 35° C. n-Heptane (240 mL) was added to the crude product and was stirred for 45 min and was filtered. The filter cake was washed three times with n-heptane (100 mL each) and dried under vacuum at 35° C. The product was obtained as an approximately equimolar mixture of diastereomers as a white solid (92.0 g, 0.200 mol). 1H NMR (300 MHz, CDCl3) δ 7.37-7.18 (m, 5H), 6.30 (d, J=31.1 Hz, 1H), 5.07 (s, 1H), 4.45-4.27 (m, 1H), 3.06 (d, J=4.5 Hz, 2H), 2.96 (dd, J=10.8, 8.3 Hz, 1H), 1.39 (s, 9H), 1.37-1.29 (m, 3H), 1.25 (d, J=4.6 Hz, 12H), 0.85 (dt, J=11.3, 5.6 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 172.7, 155.6, 136.7, 129.6, 128.9, 127.1, 83.0, 80.4, 54.8, 55.4-53.8 (m), 39.9, 38.5, 37.6, 28.5, 25. 7, 25.1, 23.4, 22.0; ESI-MS (positive) (m/z): 461, 405.
Pinacol L-phenylalanine-L-leucine boronate, HCl salt ((R,S)-[7])
A MeCN (752 mL) solution of pinacol N-BOC-L-phenylalanine-D,L-leucine boronate ([6]; 94 g, 0.204 mol) was cooled to about 0° C. HCl gas was bubbled into the reaction mixture for 4 hours. The resulting mixture was stirred at room temperature overnight and was then evaporated to provide a solid. A slurry was formed by the addition of MeCN (250 mL) which was then stirred for 2 hours and was filtered and washed with MeCN (50 mL). The solid was then dried under vacuum at 35° C. furnishing the product as a white solid (56.4 g, 0.136 mol; HPLC purity 96.0% as a 1.5:1 mixture of (R,S)-[7] and (S,S)-[7])).
As mentioned previously the key upgrade step can be accomplished using:
- A) fractional crystallisation, or
- B) a reslurry/hydrolysis, or
- C) chromatography,
- D) or combinations of any of the above three techniques
These are exemplified in the following 3 examples.
Example 2 Operation A—Diastereomeric Upgrade of [7] by Fractional Crystallisation: The First Crystallisation
[7] (35.0 g, 88.2 mmol; (R,S)4(S,S)-diastereomeric ratio=1.50:1) was dissolved in a mixture of isobutyl acetate (350 mL) and ethanol (24.5 mL) at about 75° C. The solution was slowly cooled to ambient temperature and stirred overnight. The resulting mixture was cooled to about 0° C. and stirred for one hour and then filtrated and the isolated solid was dried under vacuum at 35° C. The product was obtained as white solid (12.5 g, 31.5 mmol, HPLC purity 98.61% ((S,R)-[7]+(S,S)-[7]), (S,R)-[7]/(S,S)-[7] ratio of 4.16:1).
The Second Crystallisation
[7] (12.5 g, 31.5 mmol; (R,S)-/(S,S)-diastereomeric ratio=4.16:1) was dissolved in a mixture of isobutyl acetate (125 mL) and ethanol (12.5 mL) at about 75° C. The solution was slowly cooled to ambient temperature and stirred overnight. The resulting mixture was cooled to about 0° C. and stirred for one hour and then filtrated and the isolated solid was dried under vacuum at 35° C. The product was obtained as white solid (7.10 g, 17.9 mmol, HPLC purity 98.58% ((S,R)-[7]+(S,S)-[7]), (S,R)-[7]/(S,S)-[7] ratio of 9.97:1).
The Third Crystallisation
[7] (7.1 g, 17.9 mmol; (R,S)-/(S,S)-diastereomeric ratio=9.97:1) was dissolved in a mixture of isobutyl acetate (71 mL) and ethanol (8.0 mL) at about 75° C. The solution was slowly cooled to ambient temperature and stirred overnight. The resulting mixture was cooled to about 0° C. and stirred for one hour and then filtrated and the isolated solid was dried under vacuum at 35° C. The product was obtained as white solid (5.4 g, 13.6 mmol, HPLC purity 96.69% ((S,R)-[7]+(S,S)-[7]), (S,R)-[7]/(S,S)-[7] ratio of 17.5:1). 1H NMR (300 MHz, d6-DMSO) δ 8.71 (d, J=15.6 Hz, 1H), 8.44 (d, 3H), 7.27 (m, 5H), 4.02 (s, 1H), 3.03 (m, 2H), 2.80 (d, J=4.5 Hz, 1H), 1.45 (m, 1H), 1.30-0.95 (m, 14H), 0.97-0.57 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 168.8, 134.1, 130.2, 129.0, 127.8, 83.7, 53.4, 40.3-36.7 (m), 32.0, 29.3, 25. 5, 25.0 (m), 23.4, 22.0; ESI-MS (positive) (m/z): 361, 261.
Example 3 Operation B—Diastereomeric Upgrade of [7] by Slurrying in a Wet Solvent:
A slurry of [7] (0.5 g, 1.26 mmol; (R,S)-/(S,S)-diastereomeric ratio=1.30:1) in ethyl acetate (15 mL) containing water (0.05 g, 2.78 mmol) was stirred at room temperature. After 72 hour, a sample was isolated as a white solid by filtration of the slurry and was analysed by HPLC showing a purity 97.7% and a (R,S)-/(S,S)-diastereomeric ratio of 7.0:1.
Example 4 Operation C—Diastereomeric Upgrade of [7] by Chromatography:
[7] (1.0 g, 2.52 mmol, (R,S)-/(S,S)-diastereomeric ratio=0.83:1 was dissolved in was dissolved in 1:4 i-PrOH/DCM (5.0 mL) and was purified by silica gel column chromatography eluting with 1:10 i-PrOH/DCM. Three fractions were collection providing 96.7% HPLC purity [7] (0.60 g; (R,S)-[7]/(S,S)-[7]=0.55:1), 97.2% HPLC purity [7] (0.10 g; (R,S)-[7]/(S,S)-[7]=1.98:1), and 95.9% HPLC purity [7] (0.20 g; (R,S)-[7]/(S,S)-[7]=2.25:1), after evaporation of the eluent.
For embodiment 2, examples are provided below.
Example 5 Pinacol N-(pyrazine-2-yl-carbonyl)-L-phenylalanine-L-leucine boronate ((R,S)-[8])
To a cooled (about 0° C.) solution of 2-pyrazinecarboxylic acid (1.61 g, 13 mmol) in DMF (84.6 mL) was added DIPEA (4.74 mL), HATU (5.43 g, 14.3 mmol) and recrystallised pinacol L-phenylalanine-L-leucine boronate HCl salt ((R,S)-[7]; 5.4 g, 13.6 mol, as a 17.5:1 diastereomeric mixture of (R,S)-[7]/(S,S)-[7]). The mixture was warmed to room temperature and was then stirred at this temperature overnight. Ethyl acetate (270 mL) and a saturated aqueous solution of sodium of chloride (260 mL) were added to the reaction mixture. After mixing, the organic layer was separated and washed with a saturated aqueous solution of sodium of chloride (182 mL), then an aqueous 0.1N solution of KHSO4 (273 mL) and finally a saturated aqueous solution of NaHCO3 (182 mL). The organic layer was dried over MgSO4, filtered and evaporated at 35° C. giving the product as white solid (5.77 g, 12.4 mmol; HPLC purity 87.8% ((R,S)-[8]+(S,S)-[8]) as a 22.0:1 diastereomeric mixture of (R,S)-[8])/(S,S)-[8]). 1H NMR (300 MHz, CDCl3) δ 9.34 (d, J=1.2 Hz, 1H), 8.72 (t, J=9.9 Hz, 1H), 8.53 (dd, J=2.4, 1.6 Hz, 1H), 8.36 (d, J=8.4 Hz, 1H), 7.26 (ddd, J=10.7, 6.9, 4.8 Hz, 5H), 6.06 (s, 1H), 4.83 (dd, J=14.1, 6.8 Hz, 1H), 3.24-3.15 (m, 2H), 3.06 (dd, J=12.5, 7.4 Hz, 1H), 1.51-1.31 (m, 3H), 1.30-1.22 (s, 12H), 0.83 (t, J=6.7 Hz, 6H); ESI-MS (positive) (m/z): 467.
Bortezomib (anhydride; N-(2-pyrazine)carbonyl-L-phenylalanine-L-leucine boroxine)
1 N HCl (37.1 mL) was added dropwise into a mixture of pinacol N-(pyrazine-2-yl-carbonyl)-L-phenylalanine-L-leucine boronate ([8]; 5.77 g, 12.4 mmol as a 22.0 (R,S)-[8])/(S,S)-[8] diastereomeric mixture) and 2-methylpropaneboronic acid (1.89 g, 18.5 mmol) in MeOH (57.7 mL) and n-heptane (57.7 mL). The reaction mixture was stirred at room temperature overnight. The water layer was separated and washed twice with n-heptane (30 mL each). The water layer was concentrated at 35° C. and DCM (30 mL) was added into the residue. 2 N NaOH (36.9 mL) was added dropwise into the reaction mixture. The water layer was separated and washed twice with DCM (30 mL each). After dilution with DCM (30 mL) 1 N HCl was added dropwise until the pH of the aqueous phase was about 6. The water layer was extracted twice with DCM (30 mL each). The DCM portions were collected together and concentrated at 35° C. Ethyl acetate (46 mL) was added into the residue and concentrated. Ethyl acetate (16 mL) was added into the residue and concentrated until approximately 10% of the original volume remained. n-Heptane (46 mL) was added and the resulting solid was then filtered, washed with n-heptane (20 mL) and dried under vacuum at 35° C. The crude bortezomib was obtained as yellow solid (3.7 g, 9.63 mmol).
Purification of bortezomib (anhydride; N-(2-pyrazine)carbonyl-L-phenylalanine-L-leucine boroxine), Form C Example 6
A mixture of crude bortezomib (24.2 g, 63.0 mol, HPLC purity: 97.9%), MeCN (181.5 mL) and i-PrOH (12.1 mL) was stirred at room temperature for 4 hours. The solid was filtered and dried at 30° C. under vacuum overnight providing bortezomib anhydride Form C as a white solid (18.6 g, 48.4 mmol, yield 76.9%, HPLC purity: 99.7% with no individual impurity>0.10%).
Example 7
A mixture of crude bortezomib (1.0 g, 2.36 mmol, HPLC purity: 90.7%) and MeCN (8.0 mL) was stirred at room temperature for 6 hours. The solid was filtered and dried at 35° C. under vacuum for 17 hours providing bortezomib anhydride Form C as a white solid (0.75 g, 1.94 mmol, yield 82%, HPLC purity: 99.2%).
Representative XRDP pattern, a DVS graph, 1H NMR spectrum, IR spectrum, and DSC and TGA traces of Form C are shown in FIGS. 1, 2, 3, 4, 5 and 6, respectively.
……………………………………
![]() SYNTHESIS
SYNTHESIS
![]()
…………………………..
SYNTHESIS
![]()
…………………………
![]() WO2010146172A2
WO2010146172A2
α-substituted boronic ester (Vl).
Scheme 1
Scheme 3.
![Figure imgf000032_0001]()
According to the preferred embodiment of Scheme 4 (wherein R1, R2, R3, X and A are as defined as in the items above), a compound of formula VIII, for example about 3.8 mmol, in form of its (R)- or (S)- enantiomer or in form of a mixture of enantiomers, can be prepared by contacting a compound of formula Vl dissolved in an organic solvent, preferably THF, with a solution comprising a reagent for substituting X with a protected amino moiety, for example sodium bis(trimethylsilyl)amide (NaHMDS) or lithium bis(trimethylsilyl)amide (LiHMDS), at suitably low temperature such as -60 0C to -10 0C, more preferably – 40 0C to -30 0C, in an inert, preferably argon atmosphere. The solution is warmed to room temperature such as about 20 0C to 25 0C, and stirred for a suitable period of time, for example 1 to 15 hours, preferably for about 5 hours. Then, the reaction mixture is evaporated to dryness and the residue is subsequently dissolved in a suitable volume of n-heptane, for example about 10 ml_, washed with a suitable volume of H2O, for example about 8 ml_, and washed with a suitable volume of saturated aqueous solution of NaCI, for example about 4 mL. The organic phase is then dried over a suitable drying agent, most preferably MgSO4, filtrated and evaporated to dryness. Compound of formula Vila obtained in such a manner is subsequently further converted into the compound of formula VIII by dissolving the previously obtained residue in a suitable volume of n-heptane, for example 20 mL, and by adding a suitable amount of anhydrous acidic solution, for example anhydrous HCI solution in Et2O, at a suitably low temperature such as -100 0C to -10 0C, more preferably -70 0C to -50 0C, in an inert, preferably argon atmosphere. The reaction mixture is warmed to room temperature, such as about 20 0C to 25 0C, and finally the precipitating solid is isolated from reaction mixture by filtration and washed with Et2O to give α- amino boronic ester (VIII).
According to another embodiment of the present invention, the racemic mixture of the α-amino boronic ester (VIII) obtained above can be further separated to yield optically pure (R)- or (S)-enantiomer by methods known in the art, such as enantiomeric resolution by crystallization with chiral acids, e.g. malic acid, tartaric acid, mandelic acid, or by chiral chromatography. In the special case wherein the borolane part of compound of formula VIII is chiral, compound of formula VIII represents a diastereomer. Since diastereomers differ in their scalar characteristics, diastereomeric compounds of formula VIII can be separated without providing a chiral environment, e.g by crystallization or chromatographic methods on achiral supporters.
Enantiomers obtained in such manner can then be subjected to further synthesis steps to yield compounds of general formula X or free acids or esters or anhydrides or salts thereof
, wherein R1 is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted aryl, or substituted or unsubstituted aralkyl;
R2 and R3 independently from each other represent substituted or unsubstituted alkyl, substituted or unsubstituted aryl, or substituted or unsubstituted aralkyl, or R2 and R3 cooperatively form a part of a 5- to 10-membered fused or unfused ring, optionally a chiral 5- to 10-membered fused or unfused ring; and peptide comprises 1-6 amino acids coupled to each other by peptide bonds with optionally acylated terminal amino group, and wherein the chiral center * is in its (R) or (S) configuration.
For example, the racemic mixture of the intermediate compound of formula VIII, for example 3-methyl-1- (4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)butan-1-amine hydrochloride is first separated by enantiomeric resolution method known in the art to give (R)-3-m ethyl- 1 -(4,4,5, 5-tetram ethyl- 1 , 3,2- dioxaborolan-2-yl)butan-1-amine hydrochloride, which can then be subjected to further synthesis steps to yield bortezomib by synthesis routes known to or readily devisable by a person skilled in the art. For example, the following synthesis routes may be applied:
enantiomeric resolution coupling reagent
3-methyl-1 -(4,4,5,5-tetramethyl-
1,3,2-dioxaborolan-2-yl)butan-1-amine
hydrochloride hydrochloride
Scheme 5
Another aspect of the invention is a conversion of a compound of formula V or Vl to its trifuoroborate derivative compound of formula V* and Vl*, respectively, which can be further converted to compound VIII* as depicted in Scheme 6 below.
Scheme 6
………………………
![]() EP2377868A1
EP2377868A1
Improved process for manufacturing the proteasome inhibitor bortezomib. Thus, in one embodiment, the invention provides a large-scale process for forming a compound of formula ( XIV ):
or a boronic acid anhydride thereof. The process comprises the steps:
- (a) providing a boron “ate” complex of formula ( XV ):
wherein:
- R3
- is a nucleofugic group;
- Y
- is a nucleofugic group; and
- M+
- is an alkali metal;
- (b) contacting the boron “ate” complex of formula ( XV ) with a Lewis acid under conditions that afford a boronic ester compound of formula ( XVI ):
said contacting step being conducted in a reaction mixture comprising:
- (i) a coordinating ether solvent that has low miscibility with water; or
- (ii) an ether solvent that has low miscibility with water and a coordinating co-solvent;
- (c) treating the boronic ester compound of formula ( XVI ) with a reagent of formula M1-N(G)2, where M1 is an alkali metal and each G individually or together is an amino group protecting group, to form a compound of formula ( XVII ):
- (d) removing the G groups to form a compound of formula ( XVIII ):
or an acid addition salt thereof;
- (e) coupling the compound of formula ( XVIII ) with a compound of formula ( XIX );
wherein:
- P1 is a cleavable amino group protecting moiety; and
- X is OH or a leaving group;
to form a compound of formula ( XX ):
wherein P1 is as defined above;
- (f) removing the protecting group P1 to form a compound of formula ( XXI ):
or an acid addition salt thereof;
- (g) coupling the compound of formula ( XXI ) with a reagent of formula ( XXII )
wherein X is a OH or a leaving group, to form a compound of formula ( XXIII ):
and
- (h) deprotecting the boronic acid moiety to form the compound of formula ( XIV ) or a boronic acid anhydride thereof.
Compound 1 (1R)-(1S,2S,3R,5S)-pinanediol-1-ammoniumtrifluoroacetate-3-methylbutane-1-boronate
Compound 2(1S)-(1S,2S,3R,5S)-pinanediol-1-ammoniumtrifluoroacetate-3-methylbutane-1-boronate
SubstancesCompound 3 N-(2-Pyrazinecarbonyl)-L-phenylalanine-L-leucine boronic anhydride
Compound4N-(2-Pyrazinecarbonyl)-D-phenylalanine-L-leucine boronic anhydride
Compound 5 N-(2-Pyrazinecarbonyl)-L-phenylalanine-D-leucine boronic anhydride
N-(2-Pyrazinecarbonyl)-L-phenylalanine-L-leucine boronic anhydride
-
A solution of (1S,2S,3R,5S)-Pinanediol N-(2-pyrazinecarbonyl)-L-phenylalanine-L-leucine boronate (25.2 g) in 207 mL of MeOH and 190 mL of hexane was cooled to 15 °C, and 109.4 mL of 1N HCl were added in portions, keeping the temperature between 15 and 25 °C. 2-Methylpropaneboronic acid (8.67 g) was then added under vigorous stirring, and the stirring of the biphasic mixture was continued over night. After separation of the two phases, the lower layer was extracted once with 75 mL of hexane. The lower layer was then concentrated in vacuo until it became cloudy, followed by the addition of 109.4 mL of 2N NaOH and 100 mL of Et2O. The two phases were separated the lower layer was extracted with Et2O (4 · 100 mL each), and then brought to pH 6.0 by the addition of 109 mL of 1N HCl. After extraction with 100 mL of ethyl acetate, the lower layer was adjusted to pH 6.0 with 1N HCl and extracted one more time with 75 mL of ethyl acetate. The combined ethyl acetate layers were washed with semi-saturated brine (2 · 25 mL) and brine (2 · 25 mL), dried over Na2SO4, filtered, and concentrated to afford 15.3 g (81.8 %) of crude N-(2-Pyrazinecarbonyl)-L-phenylalanine-L-leucine boronic anhydride as a foam. The crude material was dissolved in 150 mL of ethyl acetate and concentrated in vacuo to a suspension, followed by the addition of 150 mL of MTBE. The suspension was stored between 2 and 8 °C over night, filtered, washed twice with MTBE, and dried under high vacuum, yielding 10.69 g (57.2 %) of N-(2-pyrazinecarbonyl)-L-phenylalanine-L-leucine boronic anhydride as a white solid.
………………..
![]() Synthesis
Synthesis
EP2627636A1
In an illustrative example of the first process of the present invention, the compound of formula (2) reacts with the compound of formula (6) ,wherein Rl is hydrogen, under presence of the coupling agent of formula (8A). The formed B-OH protected compound (4) is then deprotected to bortezomib.
The compound of formula (2) is a known compound. It may be prepared by processes known in the art, which generally start from (S)-pinanediol and 2-methylpropane boronic acid. The processes are disclosed, e.g., in WO 2005/097809, WO 2009/004350 and
WO 2009/036281. The compound (2) may be used per se or, preferably, as an acid addition salt. The most preferred acid addition salt is a trifluoroacetate salt as it is easily preparable and is crystalline.
The second reaction partner, the compound of formula (6), is advantageously prepared by a process, in which L-phenylalanine alkylester and/or its acid addition salt having the formula (5)
wherein Rl is a C1-C4 alkyl group and is preferably methyl group, is coupled in an inert solvent with pyrazine-2-carboxylic acid of formula (7) in the presence of a base.
According to one aspect of the present invention, the coupling reaction proceeds in a presence of the coupling agent of the formula (8) above, typically with the n-propylphosphonic anhydride of the formula (8A). In an advantageous embodiment, the inert solvent may be an aliphatic, cyclic or aromatic C5-C10 hydrocarbon or a halogenated C1-C4 aliphatic hydrocarbon. The base is advantageously a tertiary amine, e.g. N-methylmorpholine. The reaction temperature is typically from -20 to 0 °C, under which temperature the reaction time is about 2-4 hours. The amount of the coupling agent of formula (8) is advantageously from 1 to 2 molar equivalents in respect to the compound (5). The reaction progress may be monitored by a suitable analytical technique, e.g. by HPLC or GC. After the reaction has been terminated, the reaction mixture is elaborated by an extraction with water, by which rests of the coupling agent and the base are removed. The reaction product may be isolated from the organic phase by common means, e.g. by evaporation, or the organic phase may be used in the next step directly.
In the next step, the so formed intermediate of formula (6), in which Rl is a C1-C4 alkyl group, and is preferably methyl group, is hydrolysed by water to the compound of formula (6), in which Rl is hydrogen. Preferably, the hydrolysis is performed in a water miscible solvent in a presence of a base, e.g. an amine base. It is important to assure that essentially no epimerization occurs during the hydrolysis. Therefore, the conditions of hydrolysis must be very mild. In an advantageous mode, the basic hydrolysis under mild conditions may be performed in presence in lithium salts , for instance lithium chloride, lithium bromide, lithium nitrate, lithium trifluoroacetate , lithium tetrafluoroborate etc.
The hydrolysed compound is advantageously isolated from the reaction mixture after neutralization thereof, preferably by an extraction. The crude product may be precipitated in solid form from the extract, e.g. by using antisolvent, which typically is an aliphatic hydrocarbon such as hexane or heptanes. The crude solid may be isolated by filtration and optionally recrystallized from a suitable solvent or a solvent/antisolvent mixture.
Having the compound (2) and compound (6) available, the key step in making bortezomib according to process of the present invention comprises coupling, under presence of a base, the compound (6), in which Rl is hydrogen, with the compound (2), which preferably is charged as an acid addition salt and most preferably as trifluoroacetate salt, in an inert solvent, whereby the coupling agent necessary for the mutual reaction is the compound of formula (8), preferably of formula (8A). In an advantageous embodiment, the inert solvent may be an aliphatic, cyclic or aromatic C5-C10 hydrocarbon or a halogenated C1-C4 aliphatic hydrocarbon. The base is advantageously a tertiary amine, e.g. N-methylmorpholine. The reaction temperature is typically from -30 to 0 °C, preferably from -20 to -10°C, under which temperature the reaction time is about 1-2 hours. The amount of the coupling agent of formula (8) is advantageously from 1 to 2 molar equivalents in respect to the compound (2). The reaction progress may be monitored by a suitable analytical technique, e.g. by HPLC or GC. After the reaction has been terminated, the reaction mixture is elaborated by an extraction with water, by which rests of the coupling agent and the base are removed. The reaction product may be isolated from the organic phase by common means, e.g. by evaporation and may be optionally purified, e.g. by column chromatography.
Whenever useful, reaction products of any of the steps of the process may be used in the next step without isolation from the reaction mixture.
In the last step, the so formed protected bortezomib intermediate of the formula (4) is deprotected by yielding bortezomib of formula (1). Any of deprotecting procedures known in the art may be used. In particular, the transesterification step disclosed in WO 2005/097809, in which the protected bortezomib reacts with an organic boronic acid acceptor in acidic conditions, represents an useful deprotecting process. In an illustrative example of the second process of the present invention, the compound of formula (2) reacts in the first step with the L-phenylalanine compound of formula (5a), wherein R is a nitrogen protective group. The useful nitrogen protective group is a tert.butyloxycarbonyl group (tBOC), but is should be understood that other suitable nitrogen protective groups may be used as well.
![Figure imgf000013_0001]()
Similarly as in the first process of the present invention, the compound of formula (2) may be charged in the reaction as an acid addition salt, preferably the trifluoroacetate salt. The coupling reaction with the tBOC-protected compound (5a) typically proceeds in an inert solvent, in the presence of base and is, in accordance with the present invention, mediated by the action of the coupling agent of the formula (8), preferably (8A). The inert solvent is advantageously a chlorinated C1-C4 hydrocarbon or C5-C10 aliphatic, cyclic or aromatic hydrocarbon, the base is preferably a tertiary amine. The reaction conditions and workup of the reaction mixture are essentially the same as disclosed in the first process of the present invention.
In a next step, the protective group R in the so formed compound (3) is removed to yield a compound of formula (3) in which R is hydrogen. In case of the tBOC protective group, the deprotection is typically performed by treating the substrate with HC1 in ethyl acetate. After termination of the reaction, the product may be isolated from the reaction mixture by diluting with a hydrocarbon, e.g. with heptanes, and precipitating the product as a hydrochloride salt.
In the second coupling step, the compound (3), in which R is hydrogen, reacts with the 2-pyrazinecarboxylic acid of formula (7) in the presence of the coupling agent of the formula (8), preferably (8a), in an inert solvent and in the presence of a base.
……………………………………………………………..
EXAMPLES
EXAMPLE-1 : PROCESS FOR PREPARING N-[(1 S)-2-[[(1 R)-1-[(3aS,4S,6S,7aR)- hexahydro-3a,5,5-thmethyl-4,6-methano-1 ,3,2-benzodioxaborol-2-yl]-3-methyl butylamino]-2-oxo-1 -(phenylmethyl)ethyl] Pyrazinecarboxamide (FORMULA IX)
The process for preparing compound of formula IX comprises of the steps from Step a) to step h), which are individually demonstrated below:
Step-a) Preparation of 2-(2-Methylpropyl)-(3aS,4S,6S,7aR)-hexahydro-3a,5,5- trimethyl -4,6-methano-1 ,3,2-benzodioxaborole (Formula II):
To a stirred solution of isobutyl boronic acid (50.0 g) in n-heptane (250 ml) at 25.- 300C, was added (+)-Pinanediol (83.3 g) and stirred for I hour at 25-300C. To the reaction mass was added brine solution and the mixture was stirred. The layers were allowed to separate and the organic layer was concentrated under reduced pressure till no more solvent distills off to give the title compound (Formula II).
Step-b) Preparation of 2-((1 S)-1 -Chloro-3-methylbutyl)-(3aS,4S,6S,7aR)- hexahydro-3a,5,5- trimethyl-4,6-nnethano-1 ,3,2-benzodioxaborol (Formula III):
I. preparing a mixture of zinc chloride with tetrahydrofuran
II. preparing LDA mixture
III. preparing a solution of compound of formula-ll
in a solvent mixture comprising dichloromethane and water miscible ether solvent
IV. adding solution of step Il into the solution of step III followed by maintaining the solution at a temperature of about -40 to -700C
V. adding the mixture of step I into the product of step IV followed by maintaining the reaction mass at a temperature of about -40 to -700C
VI. raising the reaction temperature up to about 10°C to ambient temperature
VII. adding the aqueous acid solution VIII. separating the organic layer containing the compound of formula-Ill, and isolating the product.
I. Preparing a mixture of zinc chloride with tetrahydrofuran
Charged ZnCI2 (115 g) to tetrahydrofuran (805 ml) into a 1st Round bottom flask (R. B. flask) under nitrogen atmosphere at 25 to35°Cand the temperature of the resulting mixture was raised to 35 to 400C, maintained for 3-4 hours to give ZnCI2 solution.
II. Preparing LDA mixture
Charged diisopropyl amine (86 ml) to tetrahydrofuran (345 ml) into a 2nd R. B. flask under nitrogen atmosphere and resultant mixture was cooled to -7 to -150C, charged n-hexyl lithium to the above mixture and maintained for 30-40 minutes to give LDA mixture.
III. Preparing a solution of compound of formula-ll Compound of Formula Il (115.O g) was charged to a dichloromethane (161 ml) and tetrahydrofuran (690 ml) into a 3rd R. B. flask under nitrogen atmosphere at 25 to35°C and the mixture was cooled to -55 to -600C.
IV. Adding solution of step Il into the solution of step III Charged LDA mixture from the 2nd R. B. flask to the reaction mixture at -55 to -
600C and maintained for 30 minutes. The temperature was raised to -500C.
V. Adding the mixture of step I into the product of step IV
Charged ZnCI2 solution from the 1st R. B. flask at -45 to -500C and maintained for I hour. Vl. Raising the reaction temperature up to about 100C
The reaction mixture was warmed to 100C. VII. Adding the aqueous acid solution
Charged 10% H2SO4, stirred for 10-15 minutes and the organic layer was separated. VIII. Separating the Organic layer containing the compound of formula-Ill, and isolating the product.
The organic layer separated under step VII was subjected to next step, however, the aqueous layer was discarded.
Washed the organic layer with brine solution under stirring, till the aqueous layer reached to a pH around 6-7.
The organic layer was concentrated to isolate the compound of formula-Ill, under reduced pressure.
Step-c) Preparation of N,N-Bis(thmethylsilyl)-(1 R)-1 -[(3aS,4S,6S,7aR)- hexahydro-3a,5,5-thmethyl-4,6-methano-1 ,3,2-benzodioxaborol-2-yl]-3- methylbutylamine (Formula IV):
iv
Hexamethyldisilazane (101.3 ml) was charged to tetrahydrofuran (414 ml) under nitrogen atmosphere and the mixture was cooled to -20 to -300C. Charged n- hexyllithium slowly to the above mixture under stirring by maintaining the temperature at -20 to -30 0C. The reaction mixture was stirred for 1 -2 hours at -20 to -250C, charged compound of Formula III (138 g) to the above freshly prepared lithium HMDS in THF by maintaining the temperature at -15 to -200C. The reaction mixture was warmed to a temperature of 25-300C and maintained for 2-3 hours. Filtered the reaction mixture through silica bed and washed the bed with diisopropyl ether. The filtrate was concentrated under reduced pressure to a residue to give the title compound (Formula IV).
Step-d) Preparation of 4,6-Methano-1 ,3,2-benzodioxaborole-2-methanamine, hexahydro-3a,5,5-thmethyl-α-(2-methylpropyl)-,(αR,3aS,4S,6S,7aR)-,trifluoro acetate (Formula V):
Charged thfluoroacetic acid (129 ml) to diisopropyl ether (1980 ml) under nitrogen atmosphere at 25-300C and the reaction mass cooled to -100C. Charged compound of Formula IV (198 g) to the reaction mass slowly at -100C and maintained at the same temperature for 8 hours. The reaction mass was filtered, washed with diisopropyl ether (198 ml) and the obtained solid was slurry washed with water (1500 ml) at 25-300C. The slurry was filtered washed with water and the solid obtained was dried at 40-500C under reduced pressure for 8 hours to give 74.Og of the title compound (Formula V). Purity (by GC) = 99.54%
Step-e) Preparation of L-Phenylalanine methyl ester hydrochloride (Formula Vl):
To a stirred mixture of L-phenyl Alanine (25 g) in methanol (125 ml) at 25-300C, was charged thionyl chloride (13.2 ml) under stirring and the mixture was maintained at 55-600C for 2-3 hours. The reaction mass was cooled to 25-300C and concentrated under reduced pressure up to 2 volumes with respect to the staring material. Charged isopropyl alcohol (125 ml) to the reaction mass and concentrated up to 2 volumes with respect to the starting material. Cooled the reaction mass to 0-50C and maintained under stirring for 1-2 hours. Filtered the reaction mass, washed with isopropyl alcohol, suck dried for 30 minutes and the solid obtained was dried at 45- 500C for 3-4 hours to give 28.8 g of the title compound (Formula Vl). Chiral purity by HPLC: 100%.
Step-f) Preparation of L-Phenylalanine, N-(pyrazinylcarbonyl)-methyl ester (Formula VII):
![Figure imgf000038_0001]()
To a stirred mixture of Pyrazine-2-carboxylic acid (3.45 g) in DMF (50 ml) at 25-300C, was charged N-hydroxy succinimide (3.2 g) under stirring and was cooled to 0-50C. Charged N,N’-dicyclohexylcarbodiimide (DCC) (5.75 g) to the reaction mass at 0-5 0C and stirred for 15-20 minutes. Charged compound of Formula Vl (5.0 g) to the reaction mass at 0-50C and stirred for 15-20 minutes. Further, charged NMM (3.8 ml) to the reaction mass at 0-50C and stirred for 15-20 minutes. The reaction mixture was warmed to 25-300C and maintained under stirring for 2-3 hours. The reaction mass was filtered and the solid was separated. The filtrate obtained was diluted with ethylacetate (100 ml) and washed with demineralized water. The organic layer was washed with 1 N HCI, followed by washing with sodium bicarbonate solution. Concentrated the organic layer up to 2 volumes with respect to Formula Vl under reduced pressure and cooled to 25- 300C. Charged n-heptane (20 ml) to precipitate the compound, cooled the reaction mass to 0-50C, maintained for 1 -2 hours and filtered under vacuum. The solid obtained was dried at 40-450C for 3-4 hours to give 5.7 g of the title compound (Formula VII). Purity by HPLC: 99.73%, chiral purity by HPLC: 99.97%.
Step-g) Preparation of N-(pyrazinylcarbonyl)-L-Phenylalanine (Formula VIII):
To a stirred mixture of Formula VII (100 g) in acetone (500 ml) at 25-300C, was charged NaOH solution (obtained by dissolving 15.4 g of NaOH in 500 ml of water) and maintained at the same temperature for 30-50 minutes. Adjusted the pH of the reaction mass to 2 by using 1 N HCI and cooled the reaction mass to 0-50C. Maintained the reaction mass at 0-5 0C under stirring for 1 -2 hours, filtered under vacuum and dried the material obtained at 45-500C for 4-5 hours to give 84.4 g of the title compound (Formula VIII). Purity by HPLC: 99.94 % by weight. Chiral purity by HPLC: 100 %
Alternately, N-(pyrazinylcarbonyl)-L-Phenylalanine (Formula VIII) may also be prepared by
(a) Using ethylchloroformate according to the process as described below: A mixture of acetone (40 ml), pyrazine carboxylic acid (5 g) and thethylamine
(6.77 ml) was cooled to about -5°C to about 00C and ethylchloroformate (4.76 ml) was charged. The reaction mass was stirred for about 30 minutes. The reaction suspension was allowed to reach the temperature of about 25°C to about 300C and maintained for about 3 hours. The reaction suspension was cooled to about 00C to about 5°C. In the second flask the aqueous sodium hydroxide (1.68 g in 70 ml water) solution was cooled to about 00C to about 5°C and to that acetone (30 ml) and L-phenyl alanine (6.6 g) were added and the mixture was stirred for about 1 hour at that temperature. The reaction mass of the second flask was added to the reaction mass of the first flask at a temperature of about 00C to about 5°C and then stirred for about 2 hours followed by raising the temperature to about 25°C to about 300C. The reaction mass was further stirred for about 16 hours at a temperature of about 25°C to about 300C. Ethyl acetate (150 ml) was charged to the reaction solution and stirred for about 30 minutes. The layers were separated and 1 N hydrochloric acid (35 ml) was added to the separated aqueous layer. The reaction solution was cooled to about 00C to about 5°C and stirred for about 2 hours. The obtained suspension was filtered and the solid was washed with water (10 ml). The solid was then dried at a temperature of about 500C for about 4 hours to afford 2.6 g of title compound. Purity by HPLC: 99.2% by weight. Chiral purity by HPLC: 100% (b) or by using combination of EDCHCI, HOBt, according to the process as described below:
A mixture of pyrazine carboxylic acid (168.7 g), dimethylformamide (1.4 lit), hydroxybenzotriazole (HOBt:220 g), and N-methyl morpholine (221 ml) was cooled to a temperature of about 00C to about 5°C. EDC. hydrochloride (1 -Ethyl-3-(3- dimethylaminopropyl) carbodiimide-HCI; 278 g) was added to the reaction solution at a temperature of about 00C and stirred for about 30 minutes. L-phenylalanine methyl ester hydrochloride (240 g) obtained from above was dissolved in DMF (1 lit) and then added to the reaction mixture. N-methyl morpholine (110 ml) was added to the reaction mixture and the reaction mixture was maintained at a temperature of about 00C to about 5°C for about 1 hour. The reaction mixture was allowed to warm to the temperature to about 25°C to about 35°C and diluted with water (3.6 lit). The reaction mass was extracted with ethyl acetate (3×2.4 lit). The separated ethyl acetate layer was washed with 1 N hydrochloric acid (1.2 lit) and two layers were then separated. The organic layer was washed with saturated sodium bicarbonate solution (4.8 lit) and brine solution (2.4 lit). The organic layer was concentrated completely at a temperature of about 45°C to afford 260 g of pyrazine-2- carbonylphenylalanine methyl ester.
Pyrazine-2-carbonylphenylalanine methyl ester (5 g) was dissolved in acetone (25 ml) and stirred for about 5 minutes. Sodium hydroxide solution (701 mg of sodium hydroxide in 25 ml of water) was added to the reaction solution and stirred for about 3 hours at a temperature of about 25°C, and the pH was then adjusted withi N hydrochloric acid (11 ml) to a pH of about 2. The reaction mixture was cooled to a temperature of about 00C to about 5°C and stirred for about 1 hour. The suspension was filtered and suck dried to afford 4.0 g of pyrazine-2- carbonylphenylalanine.
Chiral purity by chiral HPLC: 100% Chemical purity by HPLC: 99.88%.
Step-h) Preparation of Formula IX:
To a stirred mixture of compound of Formula VIII (28.6 g) in dichloromethane (400 ml) at 25-300C under nitrogen atmosphere, were charged N-hydroxysuccinimide (13.3 g) and DCC (23.9 g) and stirred for 10-20 minutes. Charged compound of Formula-V (40 g) to the reaction mass was and stirred for 15-20 minutes.
Charged diisopropylethylamine (DIPEA) (27 ml) and maintained the reaction mass at 25-300C for 2-3 hours. The reaction mass was filtered and the solid was washed with dichloromethane (80 ml). The filtrate obtained was washed with 1 N HCI, followed by washing with sodium bicarbonate solution. Concentrated the organic layer up to 2 volumes with respect to Formula-V. Charged methanol (200 ml) and concentrated up to 2 volumes with respect to Formula-V. The concentrated mass obtained is the title compound (Formula IX).
Alternately, compound of Formula IX may also be prepared by using EDCHCI, and Hydroxybenzotriazole by a process as described below: N-(2-pyrazinecarbonyl)-L-phenylalanine (500 mg) was suspended in dichloromethane (10 ml) and cooled to about -5°C to about 00C. Hydroxybenzotriazole (HOBt:310 mg) was charged in to the reaction mass followed by the addition of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDCHCI, 385 mg) and stirred for 15 minutes. (1 R)-(S)-pinanediol 1 -ammonium trifluoroacetate-3-methylbutane-i -boronate (695 mg) was added to the reaction mixture and stirred for about 10 minutes at a temperature of about 5°C Diisopropyl ethyl amine (0.6 ml) was charged to the reaction mixture and stirred for about 30 minutes at a temperature of about 5°C The reaction mixture was allowed to warm to a temperature of about 25°C to about 300C and stirred for about 1 hour followed by the addition of 1 N hydrochloric acid (30 ml). The layers were separated and the organic layer was washed with 1 N hydrochloric acid (15 ml) and saturated sodium bicarbonate solution (2×30 ml). The organic layer was concentrated completely to afford title compound. Purity by HPLC: 84.98% Note that it is believed that 9.01 % measured by HPLC is Bortezomib that is formed prior to the final Bortezomib step. Thus, overall purity should be 84.98% + 9.0% or 93.99% as measured by HPLC.
EXAMPLE-2: PROCESS FOR PREPARING BORTEZOMIB (FORMULA I)
To a stirred mixture of compound of Formula IX (13.6 g) in methanol (272 ml) at 25- 300C, was charged n-heptane (272 ml), and isobutylboronic acid (3.2 g). Charged 2N HCI (272 ml) to the reaction mass under vigorous stirring and maintained the reaction mass at 25-300C for 1 -2 hours. After the completion of the reaction, separated the n-heptane layer and discarded. Charged n-heptane (272 ml * 2) to the aqueous layer and stirred vigorously for 10-15 minutes. Separated the n-heptane layer and the aqueous layer obtained was concentrated under vacuum at 35 to 480C. The aqueous layer was extracted with dichloromethane (272 ml) under vigorous stirring. The extraction process is repeated (272 ml *2) and the obtained dichloromethane layers were pooled and washed with saturated sodium bicarbonate solution, followed brine solution. The organic layer is separated, concentrated under vacuum to give 6 ml of the reaction mass and allowed to cool to 25-300C. Purity: 95.13% by HPLC.
Charged Toluene (102 ml) to the above reaction mass and stirred at 25-300C for 2-3 hours. Filtered the solid obtained under vacuum washed with 5% dichloromethane in toluene and dried at 45-500C under vacuum for 5 hours to give crude Bortezomib.
Yield: 7.O g (70%)
Purity by HPLC: 99.22%
Impurity-B by HPLC: 0.43%
Polymorphic Form: Form-B
XRD Pattern: As Illustrated in Fig -5 EXAMPLE-3: PROCESS FOR PURIFICATION OF BORTEZOMIB USING METHANOL AND WATER
Bortezomib (5.0 g, purity 99.22%) and methanol (15 ml) were taken into a round bottom flask and stirred at 25 to 35°c. Demineralized water (15 ml) was added to the obtained solution and stirred for 2 hours at a temperature of about 27°C. The reaction suspension was filtered and washed the solid with aqueous methanol (30 ml; water: methanol 1 :1 ). The obtained solid was dried at a temperature of about
500C for about 5 hours to afford 3.4 g of title compound.
Purity by HPLC: 99.57%
Impurity-B by HPLC: 0.30%
Further purification of the product obtained by reproducing the same process resulted in a Bortezomib having a purity of 99.6% by HPLC. Impurity-B by HPLC: 0.23%
Chiral Purity by HPLC: 99.83%
EXAMPLE-4: PROCESS FOR PREPARING BORTEZOMIB FOLLOWED BY PURIFICATION To a stirred mixture of compound of formula IX (68.3 g) in methanol (1.22 L) at 25- 300C, was charged n-heptane (1.36 L), and isobutyl boron ic acid (16.13 g). Charged 1 N HCI (13.6 L) to the reaction mass under stirring and maintained the reaction mass at 25-300C for 1 -2 hours. After the completion of the reaction, separated the n- heptane layer and discarded. Charged n-heptane (1.36 L * 2) to the aqueous layer and stirred vigorously for 10-15 minutes. Separated the n-heptane layer and the aqueous layer obtained was concentrated under vacuum. The aqueous layer was extracted with dichloromethane (13.6 L) under vigorous stirring. The extraction process is repeated (13.6 L *2) and the obtained dichloromethane layers were pooled and washed with saturated sodium bicarbonate solution, followed by brine solution. The organic layer was separated, concentrated under vacuum to give crude Bortezomib (47.0 g)
Purity by HPLC: 95.62% Impurity-B by HPLC: 0.59% Purification 1 : Bortezomib (25 g, Purity: 95.62%) and 5% ethylacetate in Toluene (250 ml) were taken into a round bottom flask and stirred at 25 to 350C for 2-3 hours. Filtered the solid obtained under vacuum washed with 5% ethylacetate in toluene and dried at 500C under vacuum for 5 hours to give Bortezomib.
Yield: 18.O g (72%) Purity by HPLC: 99.68% Impurity-B by HPLC: 0.27%
Purification 2: Bortezomib (18.0 g, purity 99.68%) and methanol (54 ml) were taken into a round bottom flask and stirred. Filtered the reaction mass through scinted funnel and washed the bed with 18 ml methanol. Demineralized water (72 ml) was added to the obtained filtrate and stirred for 2 hours at a temperature of about 27°C. The reaction suspension was filtered and washed the solid with aqueous methanol (108 ml; Water : methanol 1 :1 ). The obtained solid was dried at a temperature of about 500C for about 5 hours to afford 14 g of title compound.
Yield: 14.O g (77%) Purity by HPLC: 99.83%
Impurity B: 0.15% (by HPLC) Chiral Purity by HPLC: 99.85%
………………………….
Journal of Medicinal Chemistry, 2009 , vol. 52, 14 p. 4192 – 4199
Scheme 3. General Synthesis of Dipeptidyl Pinanediol Boronates and Boronic Acidsa
![]() References
References
- Takimoto CH, Calvo E. “Principles of Oncologic Pharmacotherapy” in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds) Cancer Management: A Multidisciplinary Approach. 11 ed. 2008.
- Adams J, Kauffman M (2004). “Development of the Proteasome Inhibitor Velcade (Bortezomib)”. Cancer Invest 22 (2): 304–11. doi:10.1081/CNV-120030218.PMID 15199612.
- Stephen R.Byrn et al (2011). “Analysis of two commercially available bortezomib products: differences in assay of active agent and impurity profile”. AAPS PharmSciTech (April 1).
- Bonvini P, Zorzi E, Basso G, Rosolen A (2007). “Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma”. Leukemia 21 (4): 838–42. doi:10.1038/sj.leu.2404528. PMID 17268529.
- Gelman JS, Sironi J, Berezniuk I, Dasgupta S, Castro LM, Gozzo FC, Ferro ES, Fricker LD (2013). “Alterations of the intracellular peptidome in response to the proteasome inhibitor bortezomib”. In Gartel, Andrei L. PLoS One 8 (8): e53263.doi:10.1371/journal.pone.0053263. PMC 3538785. PMID 23308178.
- Voorhees PM, Dees EC, O’Neil B, Orlowski RZ (2003). “The proteasome as a target for cancer therapy”. Clin Cancer Res 9 (17): 6316–25. PMID 14695130.
- “NHS watchdog rejects cancer drug”. BBC News UK. 20 October 2006. Retrieved 2009-08-14.
- “Summary of VELCADE Response Scheme”. Retrieved 2009-08-14.
- “More Velcade-Style Risk-Sharing In The UK?”. Euro Pharma Today. 21 January 2009. Retrieved 2009-08-14.
- Oakervee HE, Popat R, Curry N, et al. (2005). “PAD combination therapy (PS-341/bortezomib, doxorubicin and dexamethasone) for previously untreated patients with multiple myeloma”. Br J Haematol 129 (6): 755–62. doi:10.1111/j.1365-2141.2005.05519.x. PMID 15953001.
- Pour L., Adam Z., Buresova L., et al. (2009). “Varicella-zoster virus prophylaxis with low-dose acyclovir in patients with multiple myeloma treated with bortezomib”. Clinical Lymphoma & Myeloma 9 (2): 151–3. doi:10.3816/CLM.2009.n.036. PMID 19406726.
- Highlights Of Prescribing Information
- “Cancer drug benefits could be negated by healthy tea treatment”. Belfast Telegraph. 3 February 2009. Retrieved 2009-08-14.
- “Green tea may counteract anticancer effects of cancer therapy, bortezomib (Velcade)”. Retrieved 15 July 2013.
- “Green tea clash with bortezomib suggested”. Retrieved 26 July 2013.
- Golden EB, et al.; Lam, P. Y.; Kardosh, A.; Gaffney, K. J.; Cadenas, E.; Louie, S. G.; Petasis, N. A.; Chen, T. C.; Schonthal, A. H. (2009). “Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors”. Blood 113 (23): 5927–37.doi:10.1182/blood-2008-07-171389. PMID 19190249.
- Curran M, McKeage K. (2009). “Bortezomib: A Review of its Use in Patients with Multiple Myeloma”. Drugs 69 (7): 859–888. doi:10.2165/00003495-200969070-00006.PMID 19441872. doi:10.2165/00003495-200969070-00006.
- Journal of Pharmaceutical Sciences, 2000 , vol. 89, 6 p. 758 – 765
-
WO2009/36281 A2, AND
US2013/85277 A1,
| EP2377868A1 * |
Mar 24, 2005 |
Oct 19, 2011 |
Millennium Pharmaceuticals, Inc. |
Synthesis of Bortezomib |
| WO2005097809A2 |
Mar 24, 2005 |
Oct 20, 2005 |
Millennium Pharm Inc |
Synthesis of boronic ester and acid compounds |
| WO2009004350A1 |
Jul 2, 2008 |
Jan 8, 2009 |
Pliva Hrvatska D O O |
Methods for preparing bortezomib and intermediates used in its manufacture |
| WO2009006473A2 |
Jul 1, 2008 |
Jan 8, 2009 |
William W Bachovchin |
Pro-soft polypeptide proteasome inhibitors, and methods of use thereof |
| WO2009036281A2 |
Sep 12, 2008 |
Mar 19, 2009 |
Nageshwar Gunda |
Bortezomib and process for producing same |
| EP0315574A2 |
Oct 29, 1988 |
May 10, 1989 |
Hoechst Aktiengesellschaft |
Renin inhibitors |
| EP0788360B1 |
Oct 27, 1995 |
May 28, 2003 |
Millennium Pharmaceuticals, Inc. |
Boronic ester and acid compounds, synthesis and uses |
| US4924026 |
Aug 11, 1989 |
May 8, 1990 |
International Flavors & Fragrances Inc. |
Triisobutylene alcohols and esters, uses thereof in perfumery and halogenated intermediates useful for preparing same |
| US20010012907 * |
Feb 1, 2001 |
Aug 9, 2001 |
Nissan Chemical Industries, Ltd. |
Substituted cyclopentene derivatives and method for preparing the same |
| US20030008828 * |
Dec 6, 2001 |
Jan 9, 2003 |
Priestley E. Scott |
Novel lactam inhibitors of hepatitis C virus NS3 protease |
| US20070244319 |
Apr 18, 2006 |
Oct 18, 2007 |
Boaz Neil W |
Metallocenyl P-N ligands, preparation thereof, and use for asymmetric catalysis |
| WO2010146172A2 |
Jun 18, 2010 |
Dec 23, 2010 |
Lek Pharmaceuticals D.D. |
NEW SYNTHETIC ROUTE FOR THE PREPARATION OF α-AMINO BORONIC ACID DERIVATIVES VIA SUBSTITUTED ALK-1-YNES |
| WO2011098963A1 |
Feb 9, 2011 |
Aug 18, 2011 |
Ranbaxy Laboratories Limited |
Process for the preparation of bortezomib |
| WO2011099018A1 * |
Feb 15, 2010 |
Aug 18, 2011 |
Hetero Research Foundation |
Polymorphs of bortezomib |
| WO2011107912A1 |
Feb 24, 2011 |
Sep 9, 2011 |
Ranbaxy Laboratories Limited |
Polymorphic forms of bortezomib |
| WO2012048745A1 |
Oct 14, 2010 |
Apr 19, 2012 |
Synthon Bv |
Process for making bortezomib and intermediates for the process |
| CN102206188B |
Apr 8, 2011 |
Feb 27, 2013 |
苏州二叶制药有限公司 |
Preparation method for N-(pyrazine-2-yl carbonyl)-L-phenylalanine |
| CN102212036B |
Apr 8, 2011 |
Oct 17, 2012 |
上海医药工业研究院 |
Preparation method of N-(pyrazine-2-radical carbonyl)-L-phenyl alanine |
| EP2270019A1 |
Jun 19, 2009 |
Jan 5, 2011 |
LEK Pharmaceuticals d.d. |
New synthetic route for the preparation of alpha-amino boronic esters |
| EP2280016A1 |
Jul 27, 2009 |
Feb 2, 2011 |
LEK Pharmaceuticals d.d. |
New synthetic route for the preparation of alpha-amino boronic esters via substituted alk-1-ynes |
| US8497374 |
May 12, 2011 |
Jul 30, 2013 |
Scinopharm Taiwan, Ltd. |
Process for preparing and purifying bortezomib |
![]()
![]()
Filed under:
Uncategorized Tagged:
bortezomib ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()















 hyaluronan
hyaluronan
 sodium hyaluronate
sodium hyaluronate








 IBRUTINIB
IBRUTINIB





























































































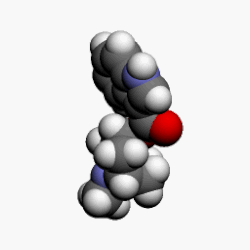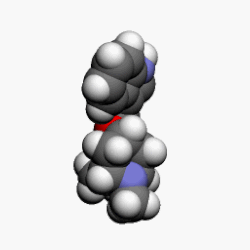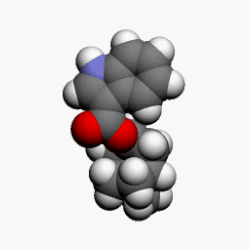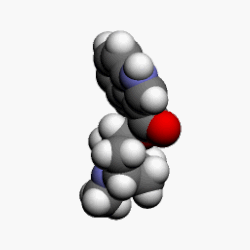






























 Originally posted on
Originally posted on 




































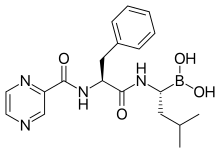



 BORTEZOMIB
BORTEZOMIB





























































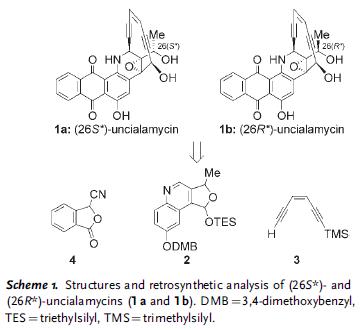

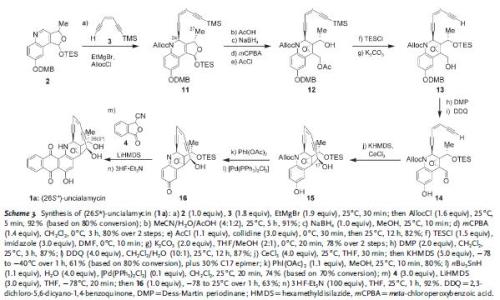
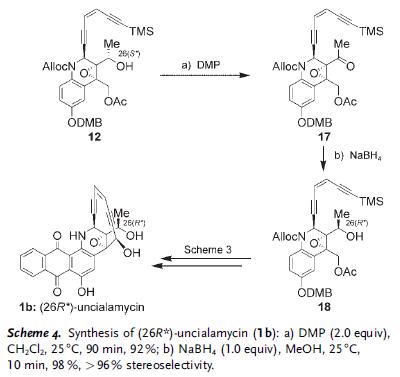




 300
300 

















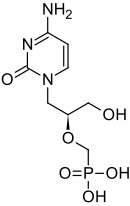






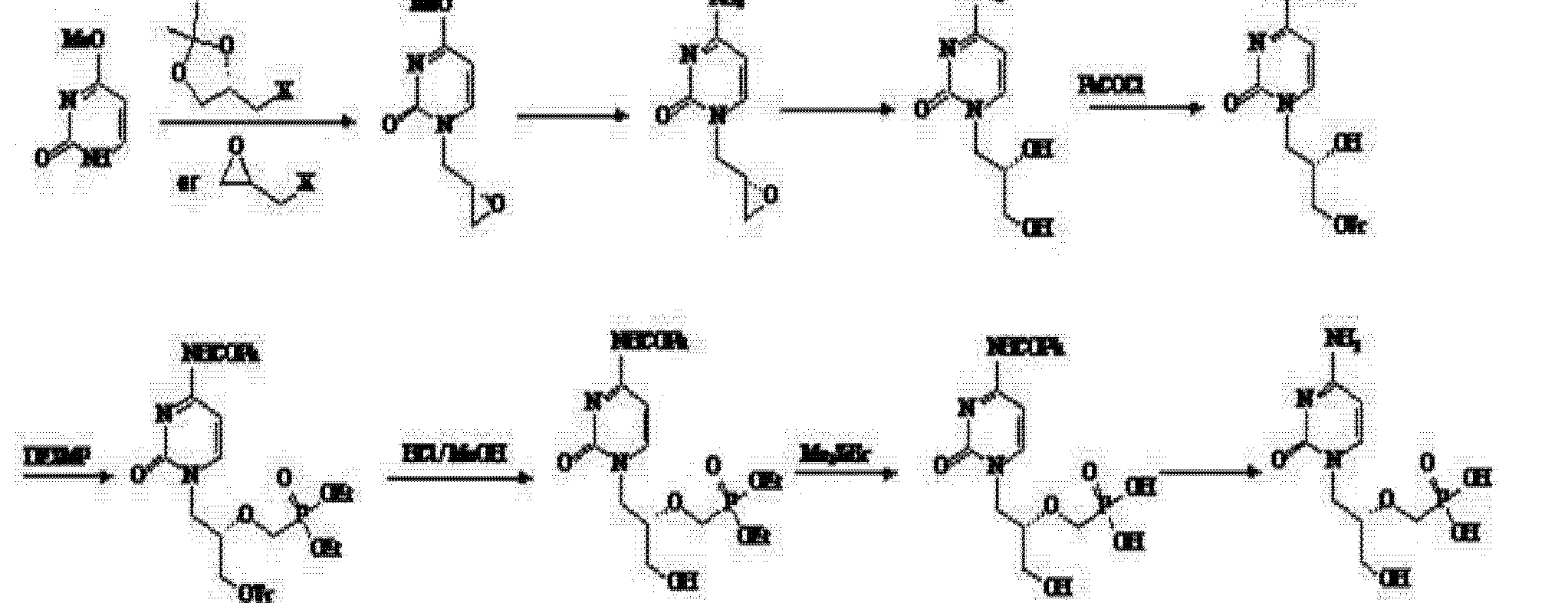
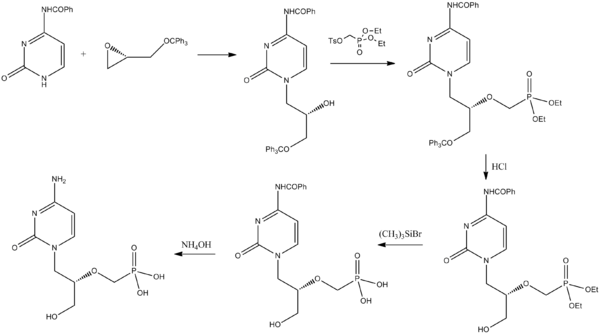











 Originally posted on
Originally posted on 






















