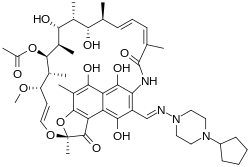
![]()
DENAGLIPTIN
(2S,4S)-1-[(2S)-2- amino-3,3-bis(4-fluorophenyl)propionyl]-4-fluoropyrrolidine-2-carbonitrile, (2S,4S)-4-fluoro-1-[4-fluoro-beta-(4-fluorophenyl)-L-phenylalanyl]-2-pyrrolidinecarbonitrile
1-[2(S)-Amino-3,3-bis(4-fluorophenyl)propionyl]-4(S)-fluoropyrrolidine-2(S)-carbonitrile
GSK-823093, 823093
811432-66-3 CAS TOSYLATE
483369-58-0 (free base)
Denagliptin (GSK-823093) having the structural formula D below is (2S,4S)-1-[(2S)-2- amino-3,3-bis(4-fluorophenyl)propionyl]-4-fluoropyrrolidine-2-carbonitrile, also named (2S,4S)-4-fluoro-1-[4-fluoro-beta-(4-fluorophenyl)-L-phenylalanyl]-2-pyrrolidinecarbonitrile
(D) – A -
Denagliptin is specifically disclosed in US Patent No. 7,132,443 and in WO 03/002531. In one embodiment, denagliptin is in the form of its hydrochloride salt as disclosed in Example 2 of WO 03/002531 or its tosylate salt as disclosed in WO 2005/009956. A class of this embodiment refers to denagliptin tosylate. Crystalline anhydrous denagliptin tosylate is disclosed in WO 2005/009956.
Denagliptin is a dipeptidyl peptidase IV (DPP-IV) inhibitor which entered phase III clinical trials in 2006 for the treatment of type 2 diabetes at GlaxoSmithKline. Development of this compound was put on hold due to unfavorable preliminary data from preclinical long-term toxicity trials.
………………………
![Figure]()
………………………..
http://www.google.com/patents/US7132443
Example 2
(2S,4S)-1-[(2S)-2-Amino-3,3-bis(4-fluorophenyl)propanoyl]-4-fluoropyrrolidine-2-carbonitrile hydrochloride
A. 3,3-Bis(4-fluorophenyl)-3-hydroxypropanoic acid.
To an anhydrous THF (80 mL) solution of n-butyl lithium (46 mL of 2.5 M, 115 mmol) at 0° C. was added dropwise diisopropylamine (11.13 g, 115 mmol) and the solution stirred for 10 minutes. Keeping the solution at 0° C., acetic acid (2.64 g, 44 mmol) was added dropwise and the mixture stirred for 10 min and it was then heated 50° C. After 30 min a heavy precipitate had formed and the solution was allowed to cool. A solution of 4,4′-diflurobenzophenone (9.6 g, 0.044 mol) in THF (50 mL, anhydrous) was added at 0° C., and the solution stirred at room temperature overnight. Water (100 mL) and diethyl ether (100 mL) were added and the aqueous layer was separated and acidified with 1M HCl to pH 3. The organics were extracted with ethyl acetate (3×200 mL) followed by drying over MgSO4. Filtration and removal of the solvent in vacuo yielded a crude white solid that could be washed with cold CHCl3 to remove trace amounts of the benzophenone. The solid was dried under high vacuum yielding 5.63 g (20.2 mmol, 46% yield) of compound A as a white solid.
1H NMR (d6-DMSO) 400 MHz δ 12.4 (s(br), 1H), 7.48–7.39 (m, 4H), 7.19–7.02 (m, 4H), 5.91 (s(br), 1H), 3.25 (s, 2H) ppm.
B. 3,3-Bis(4-fluorophenyl)acrylic acid.
To a 20% solution of sulfuric acid in acetic acid (50 mL, V/V) was compound A (5.6 g, 20.2 mmol) and the mixture stirred for 30 minutes at RT. To this solution was added H2O (500 mL) and the organics were extracted with ethyl acetate (3×150 mL) followed by drying over MgSO4. Filtration and removal of the solvent in vacuo yielded a white solid. The solid was dried under high vacuum yielding 4.97 g (19.1 mmol, 95% yield) of compound B as a white solid.
1H NMR (CDCl3) 400 MHz δ 7.27–7.21 (m, 2H), 7.19–7.13 (m, 2H), 7.10–6.95 (m, 4H), 6.26 (s, 1H) ppm.
C. 3,3-Bis(4-fluorophenyl)propanoic acid.
To a solution of compound B (2.5 g, 9.61 mmol) in ethyl acetate (250 mL) was added 10% palladium on carbon (50% w/w) and hydrogenated at 1 atmosphere of hydrogen for 12 hours. The heterogeneous solution was filtered through celite and concentrated in vacuo to provide a yellow oil. The oil was dried under high vacuum yielding 2.40 g (9.16 mmol, 95% yield) of compound C as a yellow oil.
1H NMR (d6-DMSO) 400 MHz δ 12.08 (brs, 1H), 7.40–7.30 (m, 4H), 7.15–7.05 (m, 4H), 4.45 (t, 1H, J=8.1 Hz), 3.05(d, 2H, J=8.1 Hz) ppm.
D. (4S,5R)-3-[3,3-Bis(4-fluorophenyl)propanoyl]-4-methyl-5-phenyl-1,3-oxazolidin-2-one.
To a THF (50 mL, anhydrous) containing compound C (2.0 g, 7.63 mmol) was added N,N-diisopropylethylamine (1.18 g, 9.16 mmol) and then the solution cooled to −78° C. To this solution was added trimethylacetyl chloride (0.97 g, 8.01 mmol) and the solution warmed to 0° C. over 1 hour. The cloudy mixture was filtered and the filtrate added slowly over 10 min to a solution of the lithiated (4S,5R)-(−)-4-methyl-5-phenyl-2-oxazolidinone at −78° C., which was prepared by the dropwise addition of n-butyl lithium (3.0 mL of 2.5 M, 7.63 mmol) to a THF (50 mL) solution of (4S,5R)-(−)-4-methyl-5-phenyl-2-oxazolidinone (1.35 g, 7.63 mmol) at −78° C. which had stirred for 10 min to provide the lithiated (4S,5R)-(−)-4-methyl-5-phenyl-2-oxazolidinone. The yellow mixture was warmed to 0° C. and quenched with H2O (50 mL) and extracted with diethyl ether (3×250 mL) followed by drying over MgSO4. Filtration and removal of the solvent in vacuo yielded a solid. Flash chromatography (silica gel, 20% ethyl acetate/hexanes) provided compound D. The white solid was dried under high vacuum yielding 2.31 g (5.49 mmol, 72% yield) as a white solid.
1H NMR (d6-DMSO) 400 MHz δ 7.40–7.25 (m, 9H), 7.18–7.02 (m, 4H), 5.76 (d, 1H, J=7.6 Hz), 4.65 (m, 1H), 4.58 (t, 1H, J=7.6 Hz), 3.72 (dd, 1H, J=16.8, 7.0 Hz) 3.57 (dd, 1H, J=16.8, 7.0 Hz), 0.58 (d, 3H, J=6.7 Hz) ppm.
E. (4S,5R)-3-[(2S)-2-Azido-3,3-bis(4-fluorophenyl)propanoyl]-4-methyl-5-[(1E,3Z)-1-methylhexa-1,3,5-trienyl]-1,3-oxazolidin-2-one.
To a THF (50 mL anhydrous) solution containing compound D (2.0 g, 4.75 mmol) at −78° C. was added dropwise potassium bis(trimethylsilyl)amide (10.0 mL of 0.5 M toluene solution, 4.98 mmol). After stirring for 10 min 2,4,6-triisopropylbenzenesulfonyl azide (trisyl azide) (1.84 g, 5.94 mmol) in THF (10 mL, anhydrous) was added in one portion. After 3 minutes acetic acid was added (1.31 g, 21.8 mmol) at −78° C. and then the reaction quickly warmed to 30° C. and stirred for 1 hr at that temperature generating a light yellow solution. To this solution was added H2O (100 mL) and the organics were extracted with ethyl acetate (500 mL). After washing with sat NaHCO3 (100 mL) and drying over MgSO4 the solvent was reomved in vacuo yielding a yellow oil. Column chromatography (ethyl acetate/hexanes 1:9) provided compound E as a white solid. HPLC showed a single diastereoisomer. The white solid was dried under high vacuum yielding 1.71 g (3.70 mmol, 78% yield) as a white solid.
1H NMR (CDCl3) 400 MHz δ 7.42–7.35 (m, H), 7.25–7.18 (m, H), 7.10–7.06 (m, 2H), 7.05–6.92 (m, 2H), 5.95 (d, 1H, J=10.8 Hz), 5.05 (d, 1H, J=7.1 Hz), 4.60 (d, 1H, J=10.8 Hz), 4.38 (m, 1H), 0.95 (d, 3H, J=6.8 Hz) ppm.
F. (2S)-2-Azido-3,3-bis(4-fluorophenyl)propanoic acid.
To a THF/H2O (4:1, 50 mL) solution of compound E (1.5 g, 3.25 mmol) at 0° C. was added a solution of lithium hydroxide (0.272 g, 6.49 mmol) in hydrogen peroxide (1.50 mL of 30% soln in H2O, 48.75 mmol). The mixture was stirred at 0° C. for 1 hr and then quenched with Na2SO4 (6.3 g, 50 mL of 1.0 M solution in H2O). The THF was removed in vacuo and the solution acidified to pH 1 with 6.0 M HCl at 0° C. The organics were extracted with ethyl acetate (2×200 mL) followed by drying over MgSO4. Filtration and removal of the solvent in vacuo yielded a clear oil. Column chromatography (EtOAc/hexanes/acetic acid 50:50:1) provided compound F as a white solid. The solid was dried under high vacuum yielding 0.78 g (2.60 mmol, 80% yield) as a white solid.
1H NMR (CDCl3) 400 MHz δ 9.60(s(br), 1H), 7.25–7.10 (m, 4H), 7.10–6.95 (m, 4H), 4.50 (d, 2H, J=8.6 Hz) ppm.
G. (2S)-2-Amino-3,3-bis(4-fluorophenyl)propanoic acid.
To an ethyl acetate (250 mL) solution of compound F (1.5 g, 4.95 mmol) was added 10% palladium on carbon (10% w/w) and hydrogenated at 1 atmosphere of hydrogen for 12 hr. The heterogeneous solution was filtered through celite (1 g) and the filtrate concentrated in vacuo to provide a clear oil. The oil was dried under high vacuum yielding 1.30 g (4.70 mmol, 95% yield) of compound G as a white solid.
1H NMR (d6-DMSO) 400 MHz δ 10.2(s(br), 1H), 7.38–7.27(m, 4H), 7.08–6.98 (m, 4H), 4.25 (d, 1H, J=8.3 Hz), 3.95 (d, 1H, J=8.3 Hz) ppm.
H. (2S)-2-[(tert-Butoxycarbonyl)amino]-3,3-bis(4-fluorophenyl)propanoic acid.
To a CH2Cl2 (150 mL) solution containing compound G (1.30 g, 4.69 mmol) was added triethylamine (2.37 g, 23.4 mmol) and di-tert-butyl dicarbonate (1.23 g, 5.63 mmol). After stirring for 12 hr H2O (50 mL) and CH2Cl2 (300 mL) were added and the solution acidified to pH 3 with 1.0 M HCl. Separation of the ethyl acetate layer followed by drying over MgSO4 and removal of the solvent in vacuo yielded a clear oil. The oil was dried under high vacuum yielding 1.68 g (4.4 mmol, 95% yield) of compound H as a white solid.
1H NMR (d6-DMSO) 400 MHz δ 12.4 (s(br), 1H), 7.35–7.22 (m, 4H), 7.15–6.95 (m, 4H), 4.78 (t, 1H, J=8.9 Hz), 4.25 (d, 1H, J=8.9 Hz), 3.05 (m, 1H), 1.20 (s, 3H), 1.15 (s, 6H) ppm.
I. (2S,4S)-1-[(2S)-2-(tert-Butoxycarbonyl)amino-3,3-bis(4-fluorophenyl)propanoyl]-4-fluoropyrrolidine-2-carbonitrile.
To a DMF solution (25 mL anhydrous) was compound H (1.0 g, 2.65 mmol) and HATU (1.0 g, 2.65 mmol). To this solution was added N,N-diisopropylethylamine (0.462 mL, 2.65 mmol) and after 30 min (2S, 4S)-4-fluoro-2-pyrrolidinecarbonitrile 4-methylbenzenesulfonate (0.619 g, 2.12 mmol) and additional N,N-diisopropylethylamine (0.37 mL 2.12 mmol) were added. This solution was allowed to stir at RT for 12 hr and then saturated sodium bicarbonate (100 mL) was added. The resulting gummy mixture was extracted with ethyl acetate (3×100 mL) and the organics were washed with saturated NaCl (50 mL) followed by drying over MgSO4. Filtration and removal of the solvent in vacuo yielded a clear oil. The oil was chromatographed on silica gel (hexanes/EtOAc 4:1) to provide a white solid. The solid was dried under high vacuum yielding 815 mg (1.72 mmol, 65% yield) of compound I as a white solid.
1H NMR (CDCl3) 400 MHz δ 7.38–7.32 (m, 2H), 7.21–7.15 (m, 2H), 7.12–6.98(m, 4H), 5.15 (d, 1H, J=51 Hz), 5.03 (d, 1H, J=8.9 Hz, 4.89 (d, 1H, J=11.2 Hz), 4.86 (d, 1H, J=8.9 Hz), 4.40 (d, 1H, J=11.2 Hz), 3.83 (ddd, 1H, J=36.8, 12.1, 3.7 Hz), 3.05 (d, 1H, J=12.2 Hz), 2.62 (t, 1H, J=15.3 Hz), 2.25 (m, 1H), 1.38 (s, 9H) ppm.
J. (2S,4S)-1-[(2S)-2-Amino-3,3-bis(4-fluorophenyl)propanoyl]-4-fluoropyrrolidine-2-carbonitrile hydrochloride.
To compound I (0.5 g, 1.05 mmol) was added 4.0 N HCl in 1,4-dioxane (10 mL, 40 mmol) and after 3 hr diethyl ether (100 mL) was added. The resulting precipitate was collected by filtration and after drying under high vacuum 0.41 g (1.0 mmol, 95% yield) of compound J was obtained as a white solid.
1H NMR (d6-DMSO) 400 MHz δ 8.42 (s(br), 3H), 7.72–7.66 (m, 2H), 7.38–7.32 (m, 2H), 7.25–7.19 (m, 2H), 7.06–7.0 (m, 2H), 5.38 (d, 1H, J=51 Hz), 4.91 (d, 2H, J=8.8 Hz), 4.82 (d, 1H, J=11.3 Hz), 4.41 (d, 1H, J=11.3 Hz), 3.86 (ddd, 1H, J=39.2, 12.4, 3.1 Hz), 3.45 (q, 1H, J=12.4 Hz), 2.38–2.20 (m, 2H) ppm.
…………………
PAPER
Org. Process Res. Dev., 2009, 13 (5), pp 900–906
DOI: 10.1021/op900178d
http://pubs.acs.org/doi/full/10.1021/op900178d
![Figure]()
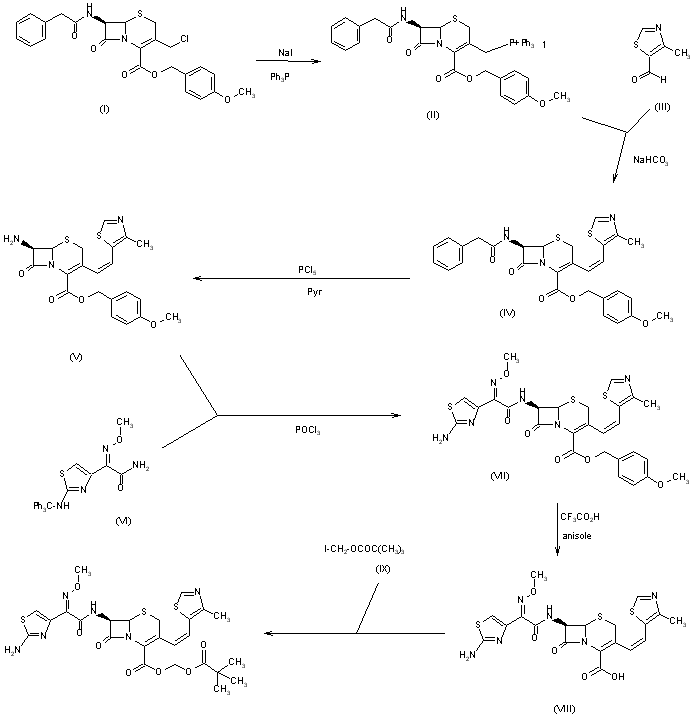
A recent paper from workers at GSK describes improvements to the synthesis of Denagliptin (12). The final chemical step is Boc deprotection of (11) with p-toluenesulphonic acid (p-TSA) in isopropanol (IPA). Some isolated batches of final product contained impurities 12A (~1%), 12B (~1%), and 12C (~0.3%). Investigation showed that these three impurities were not produced during the reaction but were produced in the dryer if there was any excess p-TSA in the filter cake during drying. These impurities could be avoided by washing the filter cake with 2 volumes of IPA prior to drying.
D.E. Paterson,* J.D. Powers, M. LeBlanc, T. Sharkey, E. Boehler, E. Irdam, and M.H. Osterhout (GlaxoSmithKline), Org. Process. Res. Dev.,2009, 13(5), 900-906.
![]()
Denagliptin Tosylate (1)
To a mixture of 11 (110 kg, 232 mmol) in isopropanol (550 L, 5 vol) at 70 °C was added a solution of p-toluenesulfonic acid monohydrate (88.4 kg, 464 mol) in isopropanol (550 L, 5 vol) over one hour while maintaining the temperature at 70 °C. After the addition, the reaction was stirred at 70 °C for 6 h. The batch was cooled to 20 °C, held for 30 min, filtered, and washed with isopropanol (2 × 220 L, 2 vol). The solids were dried at 55 °C to give 118 kg (89%) of 1 as a white solid.
Recrystallization of Denagliptin Tosylate (1)
A mixture of denagliptin tosylate (100 kg, 183 mol) and isopropanol (500 L, 5 vol) and water (500 L, 5 vol), was heated until all the solids dissolved (approximately 72 °C). The hot solution was filtered into another vessel. The solution was cooled to approximately 5 °C, and water (300 L, 3 vol) was added. The reaction was stirred at this temperature for 30 min and was filtered. The filtercake was washed with filtered isopropanol (2 × 200 L, 2 × 2 vol), and pulled dry. The solids were dried at 55 °C to give 91.9 kg (92%) of 1 as a white solid.
…………………………….
http://www.google.com.ar/patents/US7462641?cl=pt-PT
(2S,4S)-4-fluoro-1-[4-fluoro-β-(4-fluorophenyl)-L-phenylalanyl]-2-pyrrolidinecarbonitrile p-toluenesulfonic acid salt
![Figure US07462641-20081209-C00001]()
![Figure US07462641-20081209-C00003]()
EXAMPLE 1Preparation of (2S,4S)-4-fluoro-1-[4-fluoro-β-(4-fluorophenyl)-L-phenylalanyl]-2-pyrrolidinecarbonitrile p-toluenesulfonic acid salt, Form 1a) Preparation of (4S)-1-(tert-butoxycarbonyl)-4-fluoro-L-prolinamide
A reactor was charged with (4S)-1-(tert-butoxycarbonyl)-4-fluoro-L-proline (130 g, 1 wt, 1 eq.), dichloromethane (520 mL, 4 vol), pyridine (55 mL, 0.4 vol, 1.2 eq), and Boc-anhydride (145 g, 1.1 wt, 1.2 eq.). The reaction solution was stirred at approximately 20° C. for 2 hours. The reactor was charged with ammonium bicarbonate (62 g, 0.5 wt, 1.44 eq), and was stirred at approximately 20° C. overnight. The reaction was filtered over a bed of celite (130 g, 1 wt), and the filter cake was washed with dichloromethane (260 mL, 2 vol). The filtrate was concentrated to a volume of 3 volumes, heptane (520 mL, 4 vol) was added, and again concentrated to a final volume of 3 volumes. Heptane (390 mL, 3 vol) was added, and the reaction was cooled to approx. 5° C. for 30 min.
The solid was collected by filtration, washed with heptane (260 mL, 2 vol), and then dried under vacuum at approximately 50° C. to constant weight. Yield: 88-90%.
b) Preparation of (2S,4S)-4-fluoropyrrolidine-2-carbonitrile para-toluenesulfonic acid
The reactor was charged with (4S)-1-(tert-butoxycarbonyl)-4-fluoro-L-prolinamide (116 g, 1 wt, 1 eq.), isopropyl acetate (578 mL, 5 vol), and pyridine (88 mL, 0.8 vol, 2.2 eq). The resulting slurry was stirred at approx. 20° C. Trifluoroacetic anhydride (77 mL, 1.0 wt, 1.1 eq.) was added over at least 30 minutes, maintaining the temperature at approx. 20° C. The reaction solution was stirred an additonal 1 hour at approx. 20° C. Water (578 mL, 5 vol) was added slowly, and the reaction mixture was stirred for 15 minutes. The stirring was stopped, the layers were allowed to separate, and the aqueous (lower) layer was discarded. The organic layer was concentrated under vacuum at a jacket temperature of approximately 50° C. to half volume. The reaction was diluted back up to 5 volumes with isopropyl acetate. The reactor contents were cooled to 20° C., and the reactor was charged with p-toluenesulfonic acid (94 g, 0.8 wt, 1 eq). The reaction was stirred for 2 hours, and GC analysis at this point should show complete consumption of (4S)-1-(tert-butoxycarbonyl)-4-fluoro-L-prolinamide. The reaction was concentrated to 3 volumes under full vacuum at a jacket temperature of approximately 50° C. and 2 volumes of isopropyl alcohol were added. The reaction was concentrated to a final volume of 4 volumes. The reaction was cooled to 0° C. and held for 30 minutes. The solids were collected by filtration, washed with isopropyl alcohol (1 vol), and then dried under vacuum at approx. 50° C. to constant weight. Yield: 68-71%.
c) Preparation of tert-Butyl{(1S)-1-[bis(4-fluorophenyl)methyl]-2-[(2S,4S)-2-cyano-4-fluoro-1-pyrrolidinyl]-2-oxoethyl}carbamate
A reactor was charged with N-{[(1,1-dimethylethyl)oxy]carbonyl}-4-fluoro-β-(4-fluorophenyl)-L-phenylalanine (400 g, 1 wt, 1 eq.), (2S,4S)-4-fluoropyrrolidine-2-carbonitrile para-toluenesulfonic acid (307.7 g, 0.77 wt, 1.01 eq.), O-(7-Azabenzotriazol-1-yl)-N,N,N,N-tetramethyluronium hexaflurophosphate [i.e. HATU] (408 g, 1.02 wt, 1.01 equiv.), and DMF (2.8L, 7 vol). The mixture was cooled to approximately 0° C. Hunig’s base (376 mL, 0.94 vol, 2.04 equiv.) was added over at least 30 minutes. The mixture was heated to approximately 25° C. and was stirred at this temperature until the reaction was complete (ca. 3 hours). MTBE (2.8L mL, 7 vol) was added, followed by water (2L, 5 vol) over at least 30 minutes to quench the reaction. The aqueous phase was extracted with MTBE (1.2L, 3 vol). The combined organic phases were washed with water (2L, 5 vol). The organic phase was concentrated under vacuum to 3 volumes, and ethanol (1.6L, 4 vol) was added. The reaction was further concentrated under vacuum to 3 volumes, and ethanol (1.6 L, 4 vol) was added. The reaction was further concentrated under vacuum to 3 volumes. Added ethanol (2L, 5 vol). The ethanol solution of tert-Butyl {(1 S)-1-[bis(4-fluorophenyl)methyl]-2-[(2S,4S)-2-cyano-4-fluoro-1-pyrrolidinyl]-2-oxoethyl}carbamatewas used directly in the next step.
d) Preparation of (2S,4S)-4-fluoro-1-[4-fluoro-β-(4-fluorophenyl)-L-phenylalanyl]-2-pyrrolidinecarbonitrile p-toluenesulfonic acid salt. Form 1
A 10L reactor equipped with overhead stirring was charged with a slurry of tert-Butyl {(1S)-1-[bis(4-fluorophenyl)methyl]-2-[(2S,4S)-2-cyano-4-fluoro-1-pyrrolidinyl]-2-oxoethyl}carbamate (500 g, 1 wt, 1 eq) in ethanol (3.5L, 7 vol). To this solution was added para-toluenesulfonic acid (403g, 0.806 wt, 2 eq). This solution was heated to 60° C., and was allowed to stir at this temperature for 12 hours. The reaction mixture was cooled to 5° C. and was stirred at this temperature for 30 minutes. The solids were collected by filtration, washed with ethanol (2×1 L), and dried to constant weight in a 50° C. vacuum oven. Yield: 70-80% over 2 steps.
………………….
Augustyns, K. et al., “The Unique Properties of Dipeptidyl-Peptidase IV (DPP IV/CD26) and the Therapeutic Potential of DPP IV Inhibitors,” Current Medicinal Chemistry, V6, N4, 1999, pp. 311-327.
| US7132443 * |
26 Jun 2002 |
7 Nov 2006 |
Smithklinebeecham Corporation |
Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| WO2003002531A2 |
26 Jun 2002 |
9 Jan 2003 |
Curt Dale Haffner |
Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
……………………..
DIABETES
MURAGLITAZAR(CAS-No. 331741-94-7), ROSIGLITAZONE (CAS-NO. 122320-73-4), PIOGLITAZONE (CAS-No. 111025-46-8), RAGAGLITAZAR(CAS-No. 222834-30-2), FARGLITAZAR(CAS-No. 196808-45-4), TESAGLITAZAR(CAS-No. 251565-85-2), NAVEGLITAZAR(CAS-No. 476-436-68-7), NETOGLITAZONE (CAS-NO. 161600-01-7), RIVOGLITAZONE (CAS-No. 185428-18-6), K-111 (CAS-No. 221564-97-2), GW-677954 (CAS-No. 622402-24-8), FK-614 (CAS-No 193012-35-0) and (−)-Halofenate (CAS-No. 024136-23-0).
| TABLE 1 |
|
| INN or Research |
|
| Code |
Structure/Chemical Name |
|
| BIM-51077 |
L-histidyl-2-methylalanyl-L-glutamyl-glycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L- |
|
aspartyl-L-valyl-L-seryl-L-seryl-L-tyrosyl-L-leucyl-L-glutamyl-glycyl-L-glutaminyl-L-alanyl-L- |
|
alanyl-L-lysyl-L-glutamyl-L-phenylalanyl-L-isoleucyl-L-alanyl-L-tryptophyl-L-leucyl-L-valyl-L- |
|
lysyl-2-methylalanyl-L-argininamide |
| EXENATIDE |
L-histidylglycyl-L-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-aspartyl-L-leucyl- |
|
L-seryl-L-lysyl-glutaminyl-L-methionyl-L-glutamyl-L-glutamyl-L-glutamyl-L-alanyl-L-valyl-L- |
|
arginyl-L-leucyl-L-phenylalanyl-L-isoleucyl-L-glutamyl-L-tryptophyl-L-leucyl-L-lysyl-L- |
|
asparaginylglyclglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-prolyl-L- |
|
serinamide |
| CJC-1131 |
L-histidyl-D-alanyl-L-alpha-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-alpha- |
|
aspartyl-L-valyl-L-seryl-L-seryl-L-tyrosyl-L-leucyl-L-alpha-glutamylglycyl-L-glutaminyl-L-alanyl-L- |
|
alanyl-L-lysyl-L-alpha-glutamyl-L-phenylalanyl-L-isoleucyl-L-alanyl-L-tryptophyl-L-leucyl-L-valyl- |
|
L-lysylglycyl-L-arginyl-N6-[2-[2-[2-[3-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1- |
|
yl)propionamido]ethoxy]ethoxy]acetyl]-L-lysin-amide |
| LIRAGLUTIDE |
L-histidyl-L-alanyl-L-glutamyl-glycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-aspartyl-L- |
|
valyl-L-seryl-L-seryl-L-tyrosyl-L-leucyl-L-glutamyl-glycyl-L-glutaminyl-L-alanyl-L-alanyl-Nepsilon- |
|
(Nalpha-hexadecanoyl-gamma-L-glutamyl)-L-lysyl-L-glutamyl-L-phenylalanyl-L-isoleucyl-L-alanyl- |
|
L-tryptophyl-L-leucyl-L-valyl-L-arginyl-glycyl-L-arginyl-glycine |
| ZP-10 |
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe- |
|
Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-Lys-Lys-Lys-Lys-Lys-Lys-NH2 |
|
| TOLBUTAMIDE |
|
|
| TOLAZAMIDE |
|
|
| GLIPIZIDE |
|
|
| CARBUTAMIDE |
|
|
| GLISOXEPIDE |
|
|
| GLISENTIDE |
|
|
| GLIBORNURIDE |
|
|
| GLIBENCLAMIDE |
|
|
| GLIQUIDONE |
|
|
| GLIMEPIRIDE |
|
|
| GLICLAZIDE |
|
|
| METFORMIN |
|
|
| ACARBOSE |
|
|
| MIGLITOL |
|
|
| VOGLIBOSE |
|
|
| MURAGLITAZAR |
|
|
| ROSIGLITAZONE |
|
|
| PIOGLITAZONE |
|
|
| RAGAGLITAZAR |
|
|
| FARGLITAZAR |
|
|
| TESAGLITAZAR |
|
|
| NAVEGLITAZAR |
|
|
| NETOGLITAZONE |
|
|
| RIVOGLITAZONE |
|
|
| K-111 |
|
|
| GW-677954 |
|
|
| FK-614 |
|
|
| (−)-Halofenate |
|
|
| REPAGLINIDE |
|
|
| NATEGLINIDE |
|
|
| MITIGLINIDE |
|
|
| SITAGLIPTIN |
|
|
| SAXAGLIPTIN |
|
|
| VILDAGLIPTIN |
|
|
| DENAGLIPTIN |
|
|
| P32/98 |
|
|
| NVP-DPP-728 |
|
|
| SILDENAFIL |
|
|
| VARDENAFIL |
|
|
| TADALAFIL |
|
|
| PRAMLINTIDE |
L-lysyl-L-cysteinyl-L-asparaginyl-L-threonyl-L-alanyl-L-threonyl-L-cysteinyl-L-alanyl-L-threonyl- |
|
L-glutaminyl-L-arginyl-L-leucyl-L-alanyl-L-asparaginyl-L-phenylalanyl-L-leucyl-L-valyl-L-histidyl- |
|
L-seryl-L-seryl-L-asparaginyl-L-asparaginyl-L-phenylalanylglycyl-L-prolyl-L-isoleucyl-L-leucyl-L- |
|
prolyl-L-prolyl-L-threonyl-L-asparaginyl-L-valylglycyl-L-seryl-L-asparaginyl-L-threonyl-L- |
|
tyrosinamide, cyclic (2−>7)disulfide |
|
| ETOMOXIR |
|
|
| HMR-1426 |
|
|
| CETILISTAT |
|
|
| SIBUTRAMINE |
|
|
Additional information with regard to the preparation, suitable dosage forms and dose ranges of the glucagon-like-peptide-1 receptor agonists listed in Table 1 can be found in the following patents/patent applications: WO0334331, EP0981611, EP1180121, WO9808871 and WO0104156.
Filed under:
DIABETES,
Phase3 drugs,
Uncategorized Tagged:
DENAGLIPTIN,
DIABETES,
PHASE 3 ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()










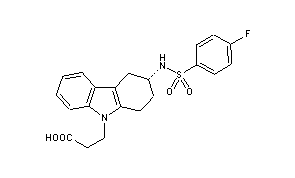
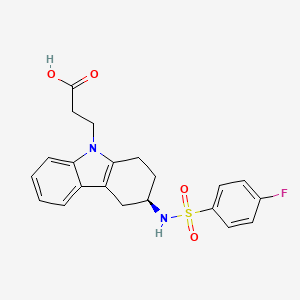
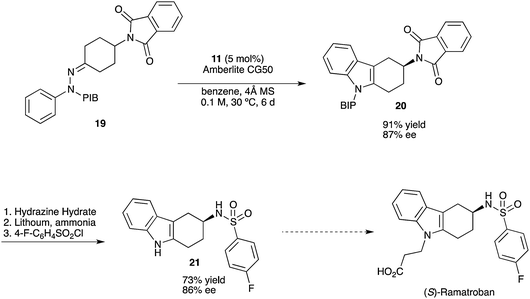
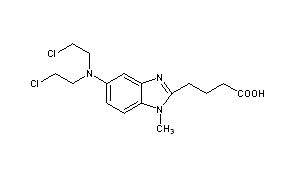
 DUTOGLIPTIN
DUTOGLIPTIN

































































































































































































































































































 Originally posted on
Originally posted on