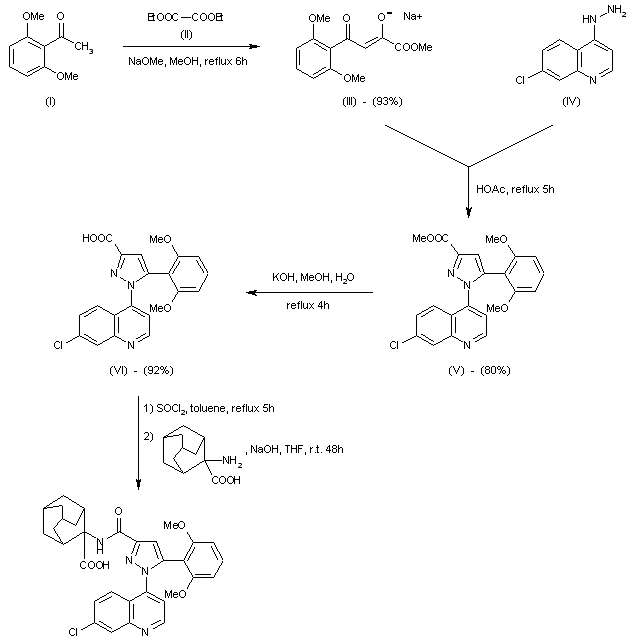
Foretinib (Exelixis, GlaxoSmithKline) (aka XL-880)WO 2012044577 A1…….Dual inhibitors of met and vegf for the treatment of castration resistant prostate cancer and osteoblastic bone metastases
or a pharmaceutically acceptable salt thereof. Compound I is known as N-(4-{[6,7- bis(methyloxy)quinolin-4-yl]oxy}phenyl)-N’-(4-fluorophenyl)cyclopropane-l, l- dicarboxamide. WO 2005/030140 describes the synthesis of N-(4-{[6,7- bis(methyloxy)quinolin-4-yl]oxy }phenyl)-N’-(4-fluorophenyl)cyclopropane-l, l- dicarboxamide (Example 12, 37, 38, and 48) and also discloses the therapeutic activity of this molecule to inhibit, regulate and/or modulate the signal transduction of kinases, (Assays, Table 4, entry 289). Example 48 is on paragraph [0353] in WO 2005/030140.
or a pharmaceutically acceptable salt thereof. Compound 2 is known as is N-[3-fluoro-4- ({6-(methyloxy)-7-[(3-morpholin-4-ylpropyl)oxy]quinolin-4-yl}oxy)phenyl]-N’-(4- fluorophenyl)cyc!opropane- 1,1 -dicarboxamide. WO 2005-030140 describes the synthesis of Compound (I) (Examples 25, 30, 36, 42, 43 and 44) and also discloses the therapeutic activity of this molecule to inhibit, regulate and/or modulate the signal transduction of kinases, (Assays, Table 4, entry 312). Compound 2 has been measured to have a c-Met IC50 value of about 0.6 nanomolar (nM). PC1YUS09/064341, which claims priority to U.S. provisional application 61/199,088, filed November 13, 2008, describes a scaled-up synthesis of Compound I.
Scheme 2
Preparation of 4-Chloro-6,7-dimethoxy-quinoIine
[00173] A reactor was charged sequentially with 6,7-dimethoxy-quinoline-4-ol (47.0 kg) and acetonitrile (318.8 kg). The resulting mixture was heated to approximately 60 °C and phosphorus oxychloride (POCl3, 130.6 kg) was added. After the addition of POCI3, the temperature of the reaction mixture was raised to approximately 77 °C. The reaction was deemed complete (approximately 13 hours) when less than 3% of the starting material remained (in-process high-performance liquid chromatography [HPLC] analysis). The reaction mixture was cooled to approximately 2-7 °C and then quenched into a chilled solution of dichloromethane (DCM, 482.8 kg), 26 percent NH4OH (251.3 kg), and water (900 L). The resulting mixture was warmed to approximately 20-25 °C, and phases were separated. The organic phase was filtered through a bed of AW hyflo super-cel NF (Celite; 5.4 kg) and the filter bed was washed with DCM (1 18.9 kg). The combined organic phase was washed with brine (282.9 kg) and mixed with water (120 L). The phases were separated and the organic phase was concentrated by vacuum distillation with the removal of solvent (approximately 95 L residual volume). DCM (686.5 kg) was charged to the reactor containing organic phase and concentrated by vacuum distillation with the removal of solvent (approximately 90 L residual volume). Methyl t-butyl ether (MTBE, 226.0 kg) was then charged and the temperature of the mixture was adjusted to -20 to -25 °C and held for 2.5 hours resulting in solid precipitate which was then filtered and washed with n-heptane (92.0 kg), and dried on a filter at approximately 25 °C under nitrogen to afford the title compound. (35.6 kg).
Preparation of -(6, 7 -Dimethoxy-quinoline- -yloxy)-phenylamine
[00174] 4-Aminophenol (24.4 kg) dissolved in N,N-dimethylacetamide (DMA, 184.3 kg) was charged to a reactor containing 4-chloro-6,7-dimethoxyquinoline (35.3 kg), sodium t- butoxide (21.4 kg) and DMA (167.2 kg) at 20-25 °C. This mixture was then heated to 100- 105 °C for approximately 13 hours. After the reaction was deemed complete as determined using in-process HPLC analysis (less than 2 percent starting material remaining), the reactor contents were cooled at 15-20 °C and water (pre-cooled, 2-7 °C, 587 L) charged at a rate to maintain 15-30 °C temperature . The resulting solid precipitate was filtered, washed with a mixture of water (47 L) and DMA (89.1 kg) and finally with water (214 L). The filter cake was then dried at approximately 25 °C on filter to yield crude 4-(6, 7-dimethoxy-quinoline-4- yloxy)-phenylamine (59.4 kg wet, 41.6 kg dry calculated based on LOD). Crude 4-(6, 7- dimethoxy-quinoline-4-yloxy)-phenylamine was refluxed (approximately 75 °C) in a mixture of tetrahydrofuran (THF, 21 1.4 kg) and DMA (108.8 kg) for approximately lhour and then cooled to 0-5 °C and aged for approximately 1 hour after which time the solid was filtered, washed with THF (147.6 kg) and dried on a filter under vacuum at approximately 25 °C to yield 4-(6,7-dimethoxy-quinoline-4-yloxy)-phenylamine (34.0 kg). Alternative Preparation of 4-(6, 7-Dimethoxy-quinoIine-4-yloxy)-phenylamine
[00175] 4-chloro-6,7-dimethoxyquinoline (34.8 kg) and 4-aminophenoI (30.8 kg) and sodium tert pentoxide (1.8 equivalents) 88.7 kg, 35 weight percent in THF) were charged to a reactor, followed by N(N-dimethylacetamide (DMA, 293.3 kg). This mixture was then heated to 105-1 15 °C for approximately 9 hours. After the reaction was deemed complete as determined using in-process HPLC analysis (less than 2 percent starting material remaining), the reactor contents were cooled at 15-25 °C and water (315 kg) was added over a two hour period while maintaining the temperature between 20-30 °C. The reaction mixture was then agitated for an additional hour at 20-25 °C. The crude product was collected by filtration and washed with a mixture of 88kg water and 82.1 kg DMA, followed by 175 kg water. The product was dried on a filter drier for 53 hours. The LOD showed less than 1 percent w/w.
[00176] In an alternative procedure, 1.6 equivalents of sodium tert-pentoxide were used and the reaction temperature was increased from 1 10-120 °C. In addition , the cool down temperature was increased to 35-40 °C and the starting temperature of the water addition was adjusted to 35-40 °C, with an allowed exotherm to 45 °C.
Preparation of l-(4-Fluoro-phenylcarbamoyl)-cyclopropanecarboxylic acid
[00177] Triethylamine (19.5 kg) was added to a cooled (approximately 5 °C) solution of cyclopropane-l,l-dicarboxylic acid (24.7 kg) in THF (89.6 kg) at a rate such that the batch temperature did not exceed 5 °C. The solution was stirred for approximately 1.3 hours, and then thionyl chloride (23.1 kg) was added, keeping the batch temperature below 10 °C. When the addition was complete, the solution was stirred for approximately 4 hours keeping temperature below 10 °C. A solution of 4-fluoroaniline (18.0 kg) in THF (33.1 kg) was then added at a rate such that the batch temperature did not exceed 10 °C. The mixture was stirred for approximately 10 hours after which the reaction was deemed complete. The reaction mixture was then diluted with isopropyl acetate (218.1 kg). This solution was washed sequentially with aqueous sodium hydroxide (10.4 kg, 50 percent dissolved in 1 19 L of water) further diluted with water (415 L), then with water (100 L) and finally with aqueous sodium chloride (20.0 kg dissolved in 100 L of water). The organic solution was concentrated by vacuum distillation (100 L residual volume) below 40 °C followed by the addition of n- heptane (171.4 kg), which resulted in the precipitation of solid. The solid was recovered by filtration and washed with n-heptane ( 102.4 kg), resulting in wet, crude l-(4-fluoro- phenylcarbamoyl)-cyclopropanecarboxylic acid (29.0 kg). The crude, l-(4-fluoro- phenylcarbamoy -cyclopropanecarboxylic acid was dissolved in methanol (139.7 kg) at approximately 25 °C followed by the addition of water (320 L) resulting in slurry which was recovered by filtration, washed sequentially with water (20 L) and n-heptane (103.1 kg) and then dried on the filter at approximately 25 °C under nitrogen to afford the title compound (25.4 kg).
Preparation of l-(4-Fluoro-phenyIcarbamoyl)-cyclopropanecarbonyl chloride
[00178] Oxalyl chloride ( 12.6 kg) was added to a solution of I -(4-fluoro- phenylcarbamoyD-cyclopropanecarboxylic acid (22.8 kg) in a mixture of THF (96.1 kg) and N, N-dimethylformamide (DMF; 0.23 kg) at a rate such that the batch temperature did not exceed 25 °C. This solution was used in the next step without further processing.
Alternative Preparation of l-(4-Fluoro-phenylcarbamoyl)-cyclopropanecarbonyl chloride
[00179] A reactor was charged with l-(4-fluoro-phenylcarbamoyl)- cyclopropanecarboxylic acid (35 kg), 344 g DMF, and 175kg THF. The reaction mixture was adjusted to 12-17 °C and then to the reaction mixture was charged 19.9 kg of oxalyl chloride over a period of 1 hour. The reaction mixture was left stirring at 12-17 °C for 3 to 8 hours. This solution was used in the next step without further processing.
Preparation of cyclopropane-l,l-dicarboxylic acid [4-(6,7-dimethoxy-quinoline-4- yloxy)-phenyl]-amide (4-fluoro-phenyl)-amide
[00180] The solution from the previous step containing l-(4-fluoro-phenylcarbamoyl)- cyclopropanecarbonyl chloride was added to a mixture of compound 4-(6,7-dimethoxy- quinoline-4-yloxy)-phenylamine (23.5 kg) and potassium carbonate (31.9 kg) in THF (245.7 kg) and water (116 L) at a rate such that the batch temperature did not exceed 30 °C. When the reaction was complete (in approximately 20 minutes), water (653 L) was added. The mixture was stirred at 20-25 °C for approximately 10 hours, which resulted in the precipitation of the product. The product was recovered by filtration, washed with a pre-made solution of THF (68.6 kg) and water (256 L), and dried first on a filter under nitrogen at approximately 25 °C and then at approximately 45 °C under vacuum to afford the title compound (41.0 kg, 38.1 kg, calculated based on LOD). Alternative Preparation of cyclopropane-l,l-dicarboxylic acid [4-(6,7-dimethoxy- quinoIine-4-yloxy)-phenyl]-amide (4-fluoro-phenyl)-amide
[00181] A reactor was charged with 4-(6,7-dimethoxy-quinoline-4-yloxy)-phenylamine (35.7 kg, 1 equivalent), followed by 412.9 kg THF. To the reaction mixture was charged a solution of 48.3 K2C03 in 169 kg water. The acid chloride solution of described in the
Alternative Preparation of l-(4-Fluoro-phenylcarbamoyl)-cvclopropanecarbonyl chloride above was transferred to the reactor containing 4-(6,7-dimethoxy-quinoline-4-yloxy)- phenylamine while maintaining the temperature between 20-30 °C over a minimum of two hours. The reaction mixture was stirred at 20-25 °C for a minimum of three hours. The reaction temperature was then adjusted to 30-25 °C and the mixture was agitated. The agitation was stopped and the phases of the mixture were allowed to separate. The lower aqueous phase was removed and discarded. To the remaining upper organic phase was added 804 kg water. The reaction was left stirring at 15-25 °C for a minimum of 16 hours.
[00182] The product precipitated. The product was filtered and washed with a mixture of 179 kg water and 157.9 kg THF in two portions. The crude product was dried under a vacuum for at least two hours. The dried product was then taken up in 285.1 kg THF. The resulting suspension was transferred to reaction vessel and agitated until the suspension became a clear (dissolved) solution, which required heating to 30-35 °C for approximately 30 minutes. 456 kg water was then added to the solution, as well as 20 kg SDAG-1 ethanol (ethanol denatured with methanol over two hours. The mixture was agitated at 15-25 °C fir at least 16 hours. The product was filtered and washed with a mixture of 143 kg water and 126.7 THF in two portions. The product was dried at a maximum temperature set point of 40 °C.
[00183] In an alternative procedure, the reaction temperature during acid chloride formation was adjusted to 10-15 °C. The recrystallization temperature was changed from 15-25 °C to 45-50 °C for 1 hour and then cooled to 15-25 °C over 2 hours.
Preparation of cyclopropane-l,l-dicarboxylic acid [4-(6,7-dimethoxy-quinoline-4- yloxy)-phenyl]-amide (4-fluoro-phenyI)-amide, malate salt
[00184] Cyclopropane- 1 , 1 -dicarboxylic acid [4-(6,7-dimethoxy-quinoline-4-yloxy)- phenyl]-amide (4-fluoro-phenyI)-amide (1-5; 13.3 kg), L-malic acid (4.96 kg), methyl ethyl ketone (MEK; 188.6 kg) and water (37.3 kg) were charged to a reactor and the mixture was heated to reflux (approximately 74 °C) for approximately 2 hours. The reactor temperature was reduced to 50 to 55 °C and the reactor contents were filtered. These sequential steps described above were repeated two more times starting with similar amounts of starting material (13.3 kg), L-Malic acid (4.96 kg), MEK (198.6 kg) and water (37.2 kg). The combined filtrate was azeotropically dried at atmospheric pressure using MEK (1 133.2 kg) (approximate residual volume 71 1 L; KF < 0.5 % w/w) at approximately 74 °C. The temperature of the reactor contents was reduced to 20 to 25 °C and held for approximately 4 hours resulting in solid precipitate which was filtered, washed with MEK (448 kg) and dried under vacuum at 50 °C to afford the title compound (45.5 kg).
Alternative Preparation of cyclopropane-l,l-dicarboxylic acid [4-(6,7-dimethoxy- quinoline-4-yIoxy)-phenyl]-amide (4-fluoro-phenyI)-amide, (L) malate salt
[00185] Cyclopropane- 1,1-dicarboxylic acid [4-(6,7-dimethoxy-quinoline-4-yloxy)- phenyl]-amide (4-fluoro-phenyI)-amide (47.9 kg), L-malic acid (17.2), 658.2 kg methyl ethyl ketone, and 129.1 kg water (37.3 kg) were charged to a reactor and the mixture was heated 50-55 °C for approximately 1-3 hours, and then at 55-60 °C for an addition al 4-5 hours. The mixture was clarified by filtration through a 1 μπι cartridge. The reactor temperature was adjusted to 20-25 °C and vacuum distilled with a vacuum at 150-200 mm Hg with a maximum jacket temperature of 55 °C to the volume range of 558-731 L.
[00186] The vacuum distillation was performed two more times with the charge of 380 kg and 380.2 kg methyl ethyl ketone, respectively. After the third distillation, the volume of the batch was adjusted to 18 v/w of cyclopropane- 1,1-dicarboxylic acid [4-(6,7-dimethoxy- quinoline-4-yloxy)-phenyl]-amide (4-fluoro-phenyI)-amide by charging 159.9 kg methyl ethyl ketone to give a total volume of 880L. An addition al vacuum distillation was carried out by adjusting 245.7 methyl ethyl ketone. The reaction mixture was left with moderate agitation at 20-25 °C for at least 24 hours. The product was filtered and washed with 415.1 kg methyl ethyl ketone in three portions. The product was dried under a vacuum with the jacket temperature set point at 45 °C.
[00187] In an alternative procedure, the order of addition was changed so that a solution of 17.7 kg L-malic acid dissolved in 129.9 kg water was added to cyclopropane- 1,1- dicarboxylic acid [4-(6,7-dimethoxy-quinoHne-4-yloxy)-phenyl]-amide (4-fluoro-phenyl)- amide (48.7 kg) in methyl ethyl ketone (673.3 kg).
Preparation of Compound 2
[00188] Compound 2 was prepared as provided in Scheme 3 and the accompanying experimental examples. Scheme 3
Toluene
[00189] In Scheme 1, Xb is Br or CI. For the names of the intermediates described within the description of Scheme 1 below, Xb is referred to as halo, wherein this halo group for these intermediates is meant to mean either Br or CI.Preparation of l-[5 methoxy-4 (3-halo propoxy)- 2 nitro-phenyl]- ethanone
[00190] Water (70 L) was charged to the solution of l-[4-(3-halo propoxy)- 3-methoxy phenyl] ethanone (both the bromo and the chloro compound are commercially available). The solution was cooled to approximately 4 °C. Concentrated sulfuric acid (129.5 kg) was added at a rate such that the batch temperature did not exceed approximately 18 °C. The resulting solution was cooled to approximately 5 °C and 70 percent nitric acid (75.8 kg) was added at a rate such that the batch temperature did not exceed approximately 10 °C. Methylene chloride, water and ice were charged to a separate reactor. The acidic reaction mixture was then added into this mixture. The methylene chloride layer was separated and the aqueous layer was back extracted with methylene chloride. The combined methylene chloride layers were washed with aqueous potassium bicarbonate solution and concentrated by vacuum distillation. 1- Butanol was added and the mixture was again concentrated by vacuum distillation. The resulting solution was stirred at approximately 20°C during which time the product crystallized. The solids were collected by filtration, washed with 1-butanol to afford compound the title compound, which was isolated as a solvent wet cake and used directly in the next step. ‘HNMR (400MHz, DMSO-d6): δ 7.69 (s, 1H), 7.24 (s, 1H); 4.23 (m, 2H), 3.94 (s, 3H), 3.78 (0-3.65 (t) (2H), 2.51 (s, 3H), 2.30-2.08 (m, 2H) LC/MS Calcd for [M(CI)+H]+ 288.1, found 288.0; Calcd for [M(Br)+H]+ 332.0, 334.0, found 331.9, 334.0.
Preparation of l-[5-methoxy-4-(3-morpholin-4-yl-propoxy)-2-nitro-phenyl]-ethanone
[00191] The solvent wet cake isolated in the previous step was dissolved in toluene. A solution of sodium iodide (67.9 kg) and potassium carbonate (83.4 kg) was added to this solution, followed by tetrabutylammonium bromide (9.92 kg) and morpholine (83.4 kg). The resulting 2 phase mixture was heated to approximately 85°C for about 9 hours. The mixture was then cooled to ambient temperature. The organic layer was removed. The aqueous layer was back extracted with toluene. The combined toluene layers were washed sequentially with two portions of saturated aqueous sodium thiosulfate followed by two portions of water. The resulting solution of the title compound was used in the next step without further processing. ‘HNMR (400MHz, DMSO-d6): δ 7.64 (s, 1 H), 7.22 (s, 1H), 4.15 (t, 2H), 3.93 (s, 3H), 3.57 (t, 4H), 2.52 (s, 3H), 2.44-2.30 (m, 6H), 1.90 (quin, 2H); LC/MS Calcd for [M+H]+ 339.2, found 339.2.
Preparation of l-[2-amino-5-methoxy-4-(3-morpholin-4-yl- propoxy)-phenyl]-ethanone
[00192] The solution from the previous step was concentrated under reduced pressure to approximately half of the original volume. Ethanol and 10 percent Pd C (50 percent water wet, 5.02 kg) were added; the resulting slurry was heated to approximately 48 °C and an aqueous solution of formic acid (22.0 kg) and potassium formate (37.0 kg) was added. When the addition was complete and the reaction deemed complete by thin layer chromatography (TLC), water was added to dissolve the by-product salts. The mixture was filtered to remove the insoluble catalyst. The filtrate was concentrated under reduced pressure and toluene was added. The mixture was made basic (pH of about 10) by the addition of aqueous potassium carbonate. The toluene layer was separated and the aqueous layer was back extracted with toluene. The combined toluene phases were dried over anhydrous sodium sulfate. The drying agent was removed by filtration and the resulting solution was used in the next step without further processing. ‘HNMR (400MHZ, DMSO-d6): δ 7.1 1 (s, 1H)„ 7.01 (br s, 2H), 6.31 (s, 1H), 3.97 (t, 2H), 3.69 (s, 3H), 3.57 (t, 4H), 2.42 (s, 3H), 2.44-2.30 (m, 6H), 1.91 (quin, 2H LC/MS Calcd for [M+H]+ 309.2, found 309.1.
Preparation of 6-methoxy-7-(3-morpholin-4-yl-propoxy)-quinoiin- 4-ol, sodium salt
[00193] A solution of sodium ethoxide (85.0 kg) in ethanol and ethyl formate (70.0 kg) was added to the solution from the previous step. The mixture was warmed to approximately 44 °C for about 3 hours. The reaction mixture was cooled to approximately 25°C. Methyl t- butyl ether (MTBE) was added which caused the product to precipitate. The product was collected by filtration and the cake was washed with MTBE and dried under reduced pressure at ambient temperature. The dried product was milled through a mesh screen to afford 60.2 kg of the title compound. ‘HNMR (400MHz, DMSO-d6): δ 1 1.22 (br s, 1H), 8.61 (d, 1H), 7.55 (s, 1H), 7.54 (s, 1H), 7.17 (d, 1H), 4.29 (t, 2 H), 3.99 (m, 2H), 3.96 (s, 3H), 3.84 (t, 2H), 3.50 (d, 2H), 3.30 (m, 2H), 3.1 1 (m, 2H), 2.35 (m, 2H), LC/MS Calcd for [M+H]+ 319.2, found 319.1.
Preparation of 4-chIor-6-methoxy-7-(3 morpholin-4-yl)-quinoline
[00194] Phosphorous oxychloride (26.32 kg) was added to a solution of 6-methoxy-7-(3- morphoIin-4-yl-propoxy)-quinolin-4-ol (5.00 kg) in acetonitrile that was heated to 50-55 °C. When the addition was complete, the mixture was heated to reflux (approximately 82 °C) and held at that temperature, with stirring for approximately 18 hours at which time it was sampled for in process HPLC analysis. The reaction was considered complete when no more than 5 percent starting material remained. The reaction mixture was then cooled to 20-25 °C and filtered to remove solids. The filtrate was then concentrated to a residue. Acetronitrile was added and the resulting solution was concentrated to a residue. Methylene chloride was added to the residue and the resulting solution was quenched with a mixture of methylene chloride and aqueous ammonium hydroxide. The resulting 2 phase mixture was separated and the aqueous layer was back extracted with methylene chloride. The combined methylene chloride solutions were dried over anhydrous magnesium sulfate, filtered and concentrated to a solid. The solids were dried at 30-40 °C under reduced pressure to afford the title compound (1.480 kg). ‘HNMR (400MHz, DMSO-d6): δ 8.61 (d, 1H), 7.56 (d, 1H), 7.45 (s, 1H), 7.38 (s, 1H), 4.21 (t, 2 H), 3.97 (s, 3H), 3.58 (m, 2H), 2.50-2.30 (m, 6H), 1.97 (quin, 2H) LC MS Calcd for [M+Hf 458.2, found 458.0.
Preparation of 4-(2-fluoro-4-nitro-phenoxy)-6-methoxy-7-(3-morphoIin-4-yl
propoxy)quinoline
[00195] A solution of 4-chIoro-6-methoxy-7-(3 morpholin-4-yl)-quinoline (2.005 kg, 5.95 mol) and 2 fluoro-4-nitrophenol (1.169 kg, 7.44 mol) in 2,6-Iutidine was heated to 140-145 °C, with stirring, for approximately 2 hours, at which time it was sampled for in process HPLC analysis. The reaction was considered complete when less than 5 percent starting materia! remained. The reaction mixture was then cooled to approximately 75 °C and water was added. Potassium carbonate was added to the mixture, which was then stirred at ambient temperature overnight. The solids that precipitated were collected by filtration, washed with aqueous potassium carbonate, and dried at 55-60 °C under reduced pressure to afford the title compound (1.7 kg). ‘HNMR (400MHz, DMSO-d6): δ 8.54 (d, 1H), 8.44 (dd, 1H), 8.18 (m, 1H), 7.60 (m, 1H), 7.43 (s, 1H), 7.42 (s, 1H), 6.75 (d, 1H), 4.19 (t, 2H), 3.90 (s, 3H), 3.56 (t, 4H), 2.44 (t, 2H), 2.36 (m, 4H), 1.96 (m, 2H). LC/MS Calcd for [M+H]+ 337.1 , 339.1 , found 337.0, 339.0.
Preparation of 3-fluoro-4-[6-methoxy-7-(3-morpholin-4-yl-propoxy)-quinolin-4-yIoxy]- phenylamine
[00196] A reactor containing 4-(2-fluoro-4-nitro-phenoxy)-6-methoxy-7-(3-morpholin-4- yl propoxy)quinoline (2.5 kg) and 10 percent palladium on carbon (50 percent water wet, 250 g) in a mixture of ethanol and water containing concentrated hydrochloric acid (1.5 L) was pressurized with hydrogen gas (approximately 40 psi). The mixture was stirred at ambient temperature. When the reaction was complete (typically 2 hours), as evidenced by in process HPLC analysis, the hydrogen was vented and the reactor inerted with argon. The reaction mixture was filtered through a bed of Celite® to remove the catalyst. Potassium carbonate was added to the filtrate until the pH of the solution was approximately 10. The resulting suspension was stirred at 20-25 °C for approximately 1 hour. The solids were collected by filtration, washed with water and dried at 50-60 °C under reduced pressure to afford the title compound (1.164 kg)._’H NMR (400MHz, DMSO-d6): δ 8.45 (d, 1H), 7.51 (s, 1H), 7.38 (s, 1H), 7.08 (t, 1H), 6.55 (dd, 1H), 6.46 (dd, 1H), 6.39 (dd, 1H), 5.51 (br. s, 2H), 4.19 (t, 2H), 3.94 (s, 3H), 3.59 (t, 4H), 2.47 (t, 2H), 2.39 (m, 4H), 1.98 (m, 2H). LC/MS Calcd for
[M+H]+ 428.2, found 428.1.
Preparation of l-(4-fluoro-phenylcarbamoyl)-cycIopropanecarboxylic acid
[00197] Triethylamine (7.78 kg) was added to a cooled (approximately 4°C) solution of commercially available cyclopropanel.l-dicarboxylic acid (9.95 kg) in THF, at a rate such that the batch temperature did not exceed 10 °C. The solution was stirred for approximately 30 minutes and then thionyl chloride (9.14 kg) was added, keeping the batch temperature below 10 °C. When the addition was complete, a solution of 4 fluoroaniline (9.4 kg) in THF was added at a rate such that the batch temperature did not exceed 10 °C. The mixture was stirred for approximately 4 hours and then diluted with isopropyl acetate. The diluted solution was washed sequentially with aqueous sodium hydroxide, water, and aqueous sodium chloride. The organic solution was concentrated by vacuum distillation. Heptane was added to the concentrate. The resulting slurry was filtered by centrifugation and the solids were dried at approximately 35 °C under vacuum to afford the title compound (10.2 kg). Ή NMR (400 MHz, DMSO-d6): δ 13.06 (br s, 1H), 10.58 (s, 1H), 7.65-7.60 (m, 2H), 7.18-7.12 (m, 2H), 1.41 (s, 4H), LC/MS Calcd for [M+H]+ 224.1 , found 224.0.
Preparation of l-(4-fluoro-phenylcarbamoyl)-cyclopropanecarbonylchloride
[00198] Oxalyl chloride (291 mL) was added slowly to a cooled (approximately 5°C) solution of l-(4-fluoro-phenylcarbamoyl)-cyclopropanecarboxylic acid in THF at a rate such that the batch temperature did not exceed 10°C. When the addition was complete, the batch was allowed to warm to ambient temperature and held with stirring for approximately 2 hours, at which time in process HPLC analysis indicated the reaction was complete. The solution was used in the next step without further processing.
Preparation of cyclopropane-l,l-dicarbox lic acid {3-fluoro-4-[6-methoxy-7-(3- morphoIin-4-yl-propoxy)-quinolin-4-ylamino]phenyl}-amide-(4 fluorophenyl)-amide
[00199] The solution from the previous step was added to a mixture of 3-fluoro-4-[6- methoxy-7-(3-mo holin-4-yl-propox )-quinolin-4-ylo y]-phenylamine (1 160 kg) and potassium carbonate (412.25 g) in THF and water at a rate such that the batch temperature was maintained at approximately 15-21 °C. When the addition was complete, the batch was warmed to ambient temperature and held with stirring for approximately 1 hour, at which time in process HPLC analysis indicated the reaction was complete. Aqueous potassium carbonate solution and isopropyl acetate were added to the batch. The resulting 2-phase mixture was stirred and then the phases were allowed to separate. The aqueous phase was back extracted with isopropyl acetate. The combined isopropyl acetate layers were washed with water followed by aqueous sodium chloride and then slurried with a mixture of magnesium sulfate and activated carbon. The slurry was filtered over Celite® and the filtrate was concentrated to an oil at approximately 30°C under vacuum to afford the title compound which was carried into the next step without further processing. Ή NMR (400MHz, DMSO- d6): δ 10.41 (s, 1H), 10.03 (s, 1H), 8.47 (d, 1H), 7.91 (dd, 1H), 7.65 (m, 2H), 7.53 (m, 2H), 7.42 (m, 2H), 7.16 (t, 2H), 6.41 (d, 1H), 4.20 (t, 2H), 3.95 (s, 3H), 3.59 (t, 4H), 2.47 (t, 2H), 2.39 (m, 4H), 1.98 (m, 2H), 1.47 (m, 4H). LC MS Calcd for [M+H]+ 633.2, found 633.1.
Preparation of the bisphosphate salt of cyclopropane-l,l-dicarboxylic acid {3-fluoro-4- [6-methoxy-7-(3-morpholin-4-yl-propoxy)-quinolin-4-ylamino]phenyl}-amide (4-fluoro- phenyl)-amide
[00200] Cyclopropane- 1,1-dicarboxy lie acid {3-fluoro-4-[6-methoxy-7-(3-morpholin-4-yl- propoxy)-quinolin-4-ylamino]phenyl}-amide-(4 fluoro phenyl)-amide from the previous step was dissolved in acetone and water. Phosphoric acid (85%, 372.48 g) was added at a rate such that the batch temperature did not exceed 30 °C. The batch was maintained at approximately 15- 30 °C with stirring for 1 hour during which time the product precipitated. The solids were collected by filtration, washed with acetone and dried at approximately 60 °C under vacuum to afford the title compound (1.533 kg). The title compound has a c-Met IC50 value of less than 50 nM. The bisphosphate salt is not shown in scheme 1. Ή NMR (400
MHz, DMSO-d6): (diphosphate) δ 10.41 (s, 1H), 10.02 (s, 1H), 8.48 (d, 1 H), 7.93 (dd, 1H), 7.65 (m, 2H), 7.53 (d, 2H), 7.42 (m, 2H), 7.17 (m, 2H), 6.48 (d, 1H), 5.6 (br s, 6H), 4.24 (t, 2H), 3.95 (s, 3H), 3.69 (bs, 4H), 2.73 (bs, 6H), 2.09 (t, 2H), 1.48 (d, 4H).
 see……
see……

























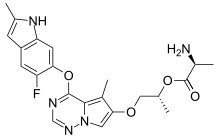 BMS-582664, brivanib alaninate
BMS-582664, brivanib alaninate
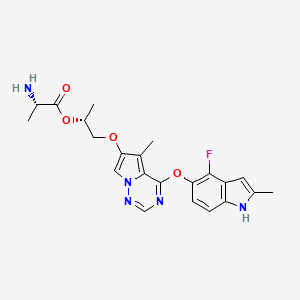
























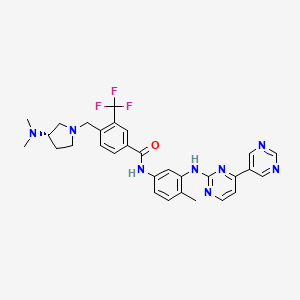
 Bafetinib
Bafetinib





















































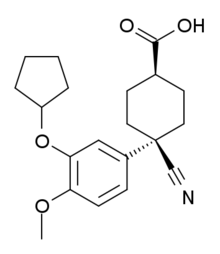
















 Cipargamin, NITD 609
Cipargamin, NITD 609






























 A practical gram scale asymmetric synthesis of CMI-977 is described. A tandem double elimination of an α-chlorooxirane and concomitant intramolecular nucleophilic substitution was used as the key step. Jacobsen hydrolytic kinetic resolution and Sharpless asymmetric epoxidation protocols were applied for the execution of the synthesis of the key chiral building block.
A practical gram scale asymmetric synthesis of CMI-977 is described. A tandem double elimination of an α-chlorooxirane and concomitant intramolecular nucleophilic substitution was used as the key step. Jacobsen hydrolytic kinetic resolution and Sharpless asymmetric epoxidation protocols were applied for the execution of the synthesis of the key chiral building block.
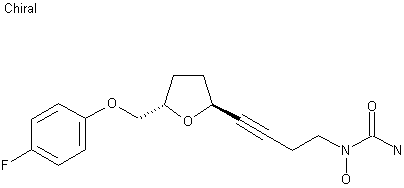
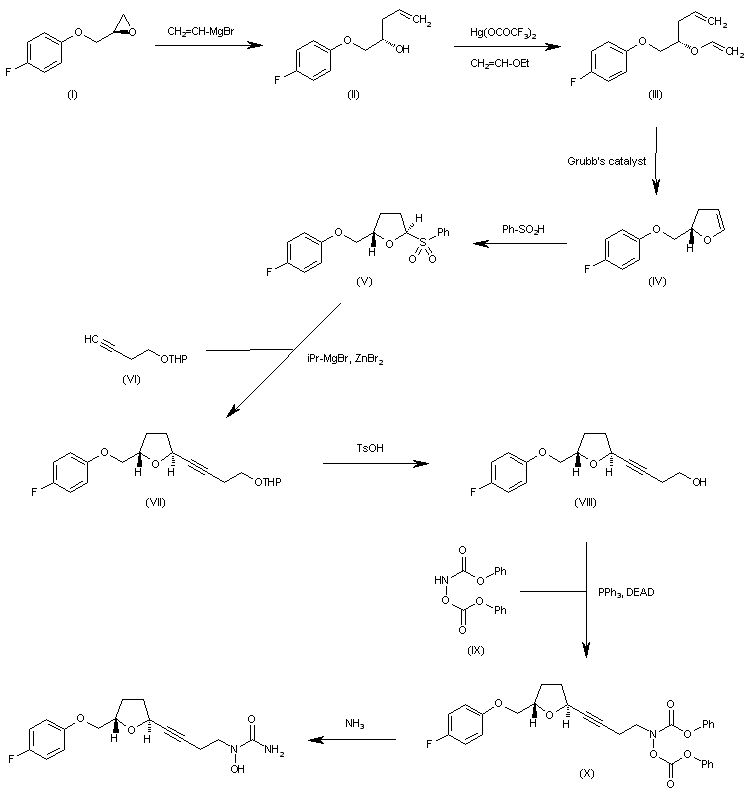 The reaction of oxirane (I) with vinylmagnesium bromide in THF gives 1-(4-fluorophenoxy)-4-penten-2(S)-ol (II), which is treated with ethyl vinyl ether and mercuric trifluoroacetate to yield the vinyl ether (III). The cyclization of (III) by means of Grubb’s catalyst in refluxing benzene affords the dihydrofuran (IV), which is treated with benzenesulfinic acid in dichloromethane to give the sulfone (V). The reaction of (V) with the acetylenic tetrahydropyranyl ether (VI) by means of isopropylmagnesium bromide in THF yields the expected addition product (VII), which is treated with TsOH to eliminate the tetrahydropyranyl group and provide the alcohol (VIII). The condensation of (VIII) with N,O-bis (phenoxycarbonyl)hydroxylamine (IX) by means of PPh3 and DEAD in THF affords the protected carbamate derivative (X), which is finally treated with ammonia in methanol.
The reaction of oxirane (I) with vinylmagnesium bromide in THF gives 1-(4-fluorophenoxy)-4-penten-2(S)-ol (II), which is treated with ethyl vinyl ether and mercuric trifluoroacetate to yield the vinyl ether (III). The cyclization of (III) by means of Grubb’s catalyst in refluxing benzene affords the dihydrofuran (IV), which is treated with benzenesulfinic acid in dichloromethane to give the sulfone (V). The reaction of (V) with the acetylenic tetrahydropyranyl ether (VI) by means of isopropylmagnesium bromide in THF yields the expected addition product (VII), which is treated with TsOH to eliminate the tetrahydropyranyl group and provide the alcohol (VIII). The condensation of (VIII) with N,O-bis (phenoxycarbonyl)hydroxylamine (IX) by means of PPh3 and DEAD in THF affords the protected carbamate derivative (X), which is finally treated with ammonia in methanol.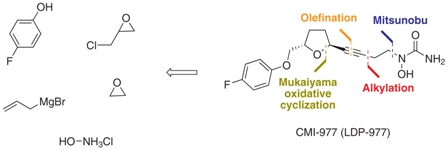

 Intermediate 4 was prepared by Cyto-Med Inc., USA, using the first synthetic route developed,5 which involved a chiral pool approach for the creation of the C9 stereogenic center (Scheme 1). A nucleophilic attack involving an oxonium electrophile intermediate, obtained from 3, produced C6, but a disappointing low degree of selectivity was observed. In a similar oxonium strategy, Ley and co-workers7 employed an anomeric oxygen to promote the carbon rearrangement of an alkynyltributylstannane to access the THF unit, but their reaction also exhibited low selectivity (Scheme 1). Other similar strategies have led to similar results.8 Gurjar et al.9 reported a new stereoselective approach that installs the stereocenters at C6 and C9 in 6 using both Jacobsen hydrolytic kinetic resolution (HKR) and a Sharpless asymmetric epoxidation step (Scheme 1). The formation of a tandem propargyl alkoxide followed by intramolecular substitution resulted in the creation of the key tetrahydrofuran ring intermediate 7. Ley and co-workers10 also explored a similar tandem strategy providing the Retrosynthetic analysis of CMI-977 (LDP-977) (1) suitable intermediate 11, which in turn afforded the key fragment 7. These two new approaches were clearly Our disconnection approach began with a superior for the construction of the 2,5-anti THF unit as higher levels of diastereoselectivity were achieved. However, numerous steps are involved in these synthetic epoxide routes. In this paper, it is described our approach for the total synthesis of CMI-977 (LDP-977) (1). The biological importance of the target molecule and its structural features inspired us to devise a more concise and diastereoselective route to achieve the THF-2,5-trans ring of intermediate 7.
Intermediate 4 was prepared by Cyto-Med Inc., USA, using the first synthetic route developed,5 which involved a chiral pool approach for the creation of the C9 stereogenic center (Scheme 1). A nucleophilic attack involving an oxonium electrophile intermediate, obtained from 3, produced C6, but a disappointing low degree of selectivity was observed. In a similar oxonium strategy, Ley and co-workers7 employed an anomeric oxygen to promote the carbon rearrangement of an alkynyltributylstannane to access the THF unit, but their reaction also exhibited low selectivity (Scheme 1). Other similar strategies have led to similar results.8 Gurjar et al.9 reported a new stereoselective approach that installs the stereocenters at C6 and C9 in 6 using both Jacobsen hydrolytic kinetic resolution (HKR) and a Sharpless asymmetric epoxidation step (Scheme 1). The formation of a tandem propargyl alkoxide followed by intramolecular substitution resulted in the creation of the key tetrahydrofuran ring intermediate 7. Ley and co-workers10 also explored a similar tandem strategy providing the Retrosynthetic analysis of CMI-977 (LDP-977) (1) suitable intermediate 11, which in turn afforded the key fragment 7. These two new approaches were clearly Our disconnection approach began with a superior for the construction of the 2,5-anti THF unit as higher levels of diastereoselectivity were achieved. However, numerous steps are involved in these synthetic epoxide routes. In this paper, it is described our approach for the total synthesis of CMI-977 (LDP-977) (1). The biological importance of the target molecule and its structural features inspired us to devise a more concise and diastereoselective route to achieve the THF-2,5-trans ring of intermediate 7.  The epoxide rac-5was resolved by hydrolytic kinetic resolution under Jacobsen conditions,14 using the catalyst (R, R)-(salen)CoIII(OAc) (19, 0.5 mol%) and H2O (0.57 equiv) in tert-butyl methyl ether, providing (S)-5 in a 48% yield.9 The next step involved the epoxide ring-opening of (S)-5 with allylmagnesium bromide (18), providing homoallylic alcohol 15 in a quantitative yield (Scheme 4).
The epoxide rac-5was resolved by hydrolytic kinetic resolution under Jacobsen conditions,14 using the catalyst (R, R)-(salen)CoIII(OAc) (19, 0.5 mol%) and H2O (0.57 equiv) in tert-butyl methyl ether, providing (S)-5 in a 48% yield.9 The next step involved the epoxide ring-opening of (S)-5 with allylmagnesium bromide (18), providing homoallylic alcohol 15 in a quantitative yield (Scheme 4). The subsequent oxidative cyclization of 15 according to the Mukaiyama protocol,12 mediated by the Co(modp)2 (20) (30 mol%) catalyst,15 provided trans-THF 14 as the only observed diastereoisomer in an 84% yield.8 This approach has proven to be a powerful strategy for accessing the 2,5-trans-THF unit in a highly diastereoselective fashion. Preparation of the key fragment 4 and conclusion of the synthesis The alcohol 14 was then oxidized to aldehyde 21 under Parikh-Doering conditions, followed by Seyferth-Gilbert homologation16 using the Ohira-Bestmann reagent 22,11 assembling the terminal acetylene 7 in a 75% yield over two steps (Scheme 5).
The subsequent oxidative cyclization of 15 according to the Mukaiyama protocol,12 mediated by the Co(modp)2 (20) (30 mol%) catalyst,15 provided trans-THF 14 as the only observed diastereoisomer in an 84% yield.8 This approach has proven to be a powerful strategy for accessing the 2,5-trans-THF unit in a highly diastereoselective fashion. Preparation of the key fragment 4 and conclusion of the synthesis The alcohol 14 was then oxidized to aldehyde 21 under Parikh-Doering conditions, followed by Seyferth-Gilbert homologation16 using the Ohira-Bestmann reagent 22,11 assembling the terminal acetylene 7 in a 75% yield over two steps (Scheme 5). The 1H NMR and 13C NMR spectra and the optical rotation of trans-THF 7 matched the reported values for this compound.9 Next, the treatment of 7 with n-BuLi and ethylene oxide 13 led to alcohol 4 in a 70% yield. As shown in Scheme 5, the preparation of hydroxycarbamate 26 (53% yield), followed by its acetylation using acetyl chloride 27, provided 12 in a quantitative yield. A Mitsunobu-like reaction between alcohol 4 and N-hydroxycarbamate 12 provided 23 in a 93% yield. Finally, 23 was ammonolysed with NH3·MeOH, yielding CMI-977 as a white solid in a 38% yield. The spectral and physical data of the synthetic sample were in complete agreement with those reported in the literature.5,7-9
The 1H NMR and 13C NMR spectra and the optical rotation of trans-THF 7 matched the reported values for this compound.9 Next, the treatment of 7 with n-BuLi and ethylene oxide 13 led to alcohol 4 in a 70% yield. As shown in Scheme 5, the preparation of hydroxycarbamate 26 (53% yield), followed by its acetylation using acetyl chloride 27, provided 12 in a quantitative yield. A Mitsunobu-like reaction between alcohol 4 and N-hydroxycarbamate 12 provided 23 in a 93% yield. Finally, 23 was ammonolysed with NH3·MeOH, yielding CMI-977 as a white solid in a 38% yield. The spectral and physical data of the synthetic sample were in complete agreement with those reported in the literature.5,7-9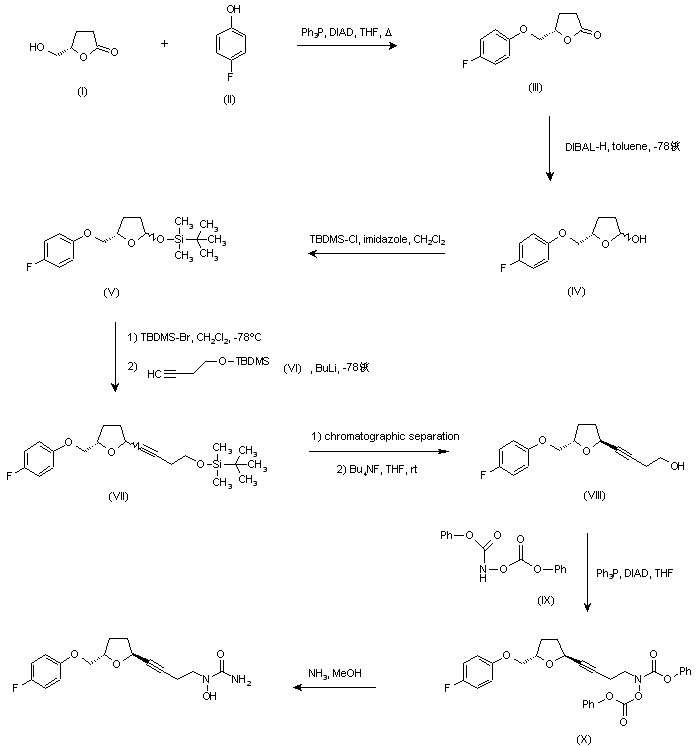 US 5703093; US 5792776; WO 9600212 Ether (III) was prepared by condensation of (S)-4-(hydroxymethyl)butyrolactone (I) and 4-fluorophenol (II) in the presence of diisopropylazodicarboxylate (DIAD) and triphenylphosphine under Mitsunobu conditions. Then, reduction of lactone (III) with DIBAL-H in toluene at -78 C gave lactol (IV), which was converted to silyl ether (V) by treatment with tert-butyldimethylsilyl chloride (TBDMS-Cl) and imidazole. Subsequent reaction of (V) with TBDMS-Br in CH2Cl2 at -78 C, followed by condensation with the lithium acetylide derived from acetylene (VI), yielded compound (VII) as a mixture of isomers. Chromatographic separation of the mixture provided the desired trans isomer, which was deprotected by treatment with tetra-n-butylammonium fluoride to give alcohol (VIII). This was then condensed with N,O-bis(phenoxycarbonyl)hydroxylamine (IX) in the presence of DIAD and Ph3P to furnish the hydroxamic acid derivative (X). Finally, concomitant deprotection of the O-phenoxycarbonyl group and substitution of the remaining phenoxy group for an amino group by treatment with methanolic ammonia in a pressure tube, provided the title compound.
US 5703093; US 5792776; WO 9600212 Ether (III) was prepared by condensation of (S)-4-(hydroxymethyl)butyrolactone (I) and 4-fluorophenol (II) in the presence of diisopropylazodicarboxylate (DIAD) and triphenylphosphine under Mitsunobu conditions. Then, reduction of lactone (III) with DIBAL-H in toluene at -78 C gave lactol (IV), which was converted to silyl ether (V) by treatment with tert-butyldimethylsilyl chloride (TBDMS-Cl) and imidazole. Subsequent reaction of (V) with TBDMS-Br in CH2Cl2 at -78 C, followed by condensation with the lithium acetylide derived from acetylene (VI), yielded compound (VII) as a mixture of isomers. Chromatographic separation of the mixture provided the desired trans isomer, which was deprotected by treatment with tetra-n-butylammonium fluoride to give alcohol (VIII). This was then condensed with N,O-bis(phenoxycarbonyl)hydroxylamine (IX) in the presence of DIAD and Ph3P to furnish the hydroxamic acid derivative (X). Finally, concomitant deprotection of the O-phenoxycarbonyl group and substitution of the remaining phenoxy group for an amino group by treatment with methanolic ammonia in a pressure tube, provided the title compound.










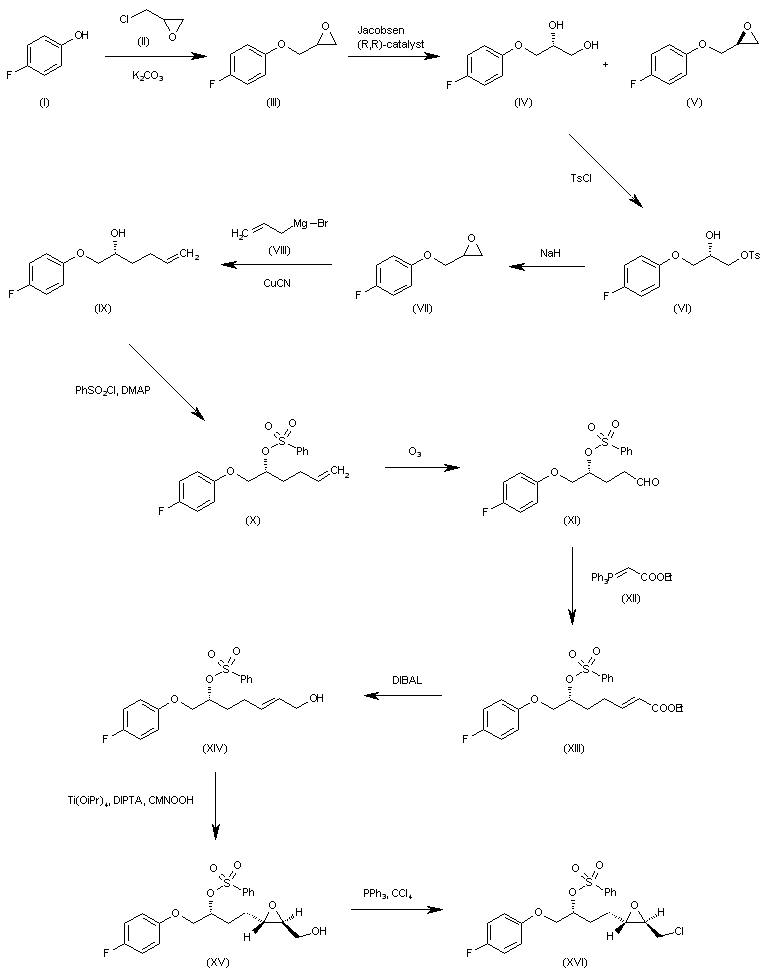 WO 0001381 The reaction of 4-fluorophenol (I) with epichlorohydrin (II) by means of K2CO3 in refluxing acetone gives 2-(4-fluorophenoxymethyl)oxirane (III), which is submitted to an enantioselective ring opening with the Jacobsen (R,R)-catalyst yielding a mixture of the (R)-diol (IV) and unaltered epoxide (V), easily separated by column chromatography. The reaction of (IV) with tosyl chloride and pyridine in dichloromethane affords the primary monotosylate (VI), which is converted into the chiral epoxide (VII) by reaction with NaH in THF/DMF. The reaction of (VII) with allylmagnesium bromide (VIII) in ethyl ether gives the 2-hexenol derivative (IX), which is treated with benzenesulfonyl chloride and DMAP yielding the sulfonate (X). The ozonolysis of (X) with ozone in dichloromethane affords the aldehyde (XI), which is condensed with ethoxycarbonylmethylene(triphenyl)phosphorane (XII) yielding the 2-heptenoic ester (XIII). The reduction of (XIII) with diisobutylaluminum hydride (DIBAL) in toluene/dichloromethane provides the 2-hepten-1-ol (XIV), which is epoxidized with cumene hydroperoxide in the presence of diisopropyl (+)-tartrate and Ti(Oi-Pr)4 in dichloromethane to give the chiral epoxyalcohol (XV). The reaction of (XV) with triphenylphosphine/CCl4 in chloroform affords the corresponding chloride (XVI). …………………………………….
WO 0001381 The reaction of 4-fluorophenol (I) with epichlorohydrin (II) by means of K2CO3 in refluxing acetone gives 2-(4-fluorophenoxymethyl)oxirane (III), which is submitted to an enantioselective ring opening with the Jacobsen (R,R)-catalyst yielding a mixture of the (R)-diol (IV) and unaltered epoxide (V), easily separated by column chromatography. The reaction of (IV) with tosyl chloride and pyridine in dichloromethane affords the primary monotosylate (VI), which is converted into the chiral epoxide (VII) by reaction with NaH in THF/DMF. The reaction of (VII) with allylmagnesium bromide (VIII) in ethyl ether gives the 2-hexenol derivative (IX), which is treated with benzenesulfonyl chloride and DMAP yielding the sulfonate (X). The ozonolysis of (X) with ozone in dichloromethane affords the aldehyde (XI), which is condensed with ethoxycarbonylmethylene(triphenyl)phosphorane (XII) yielding the 2-heptenoic ester (XIII). The reduction of (XIII) with diisobutylaluminum hydride (DIBAL) in toluene/dichloromethane provides the 2-hepten-1-ol (XIV), which is epoxidized with cumene hydroperoxide in the presence of diisopropyl (+)-tartrate and Ti(Oi-Pr)4 in dichloromethane to give the chiral epoxyalcohol (XV). The reaction of (XV) with triphenylphosphine/CCl4 in chloroform affords the corresponding chloride (XVI). …………………………………….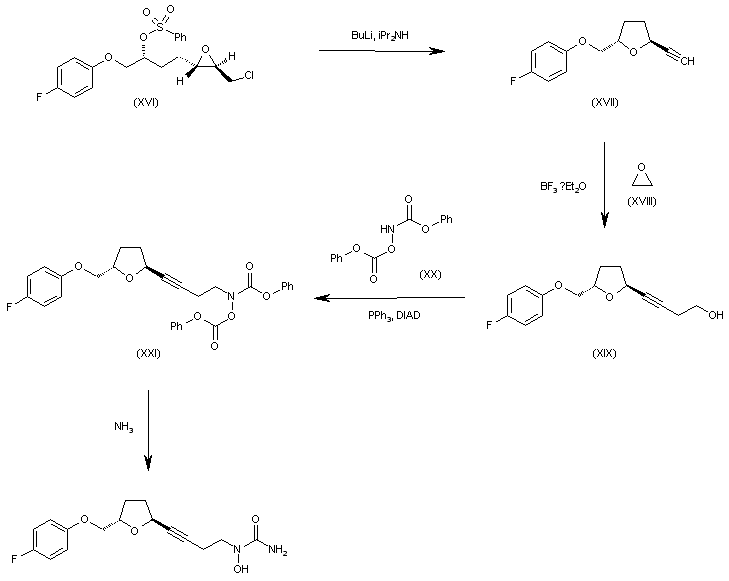 WO 0001381 Intermediate (XVI) is treated with BuLi and diisopropylamine in THF giving the chiral acetylenic tetrahydrofuran (XVII). The addition of ethylene oxide (XVIII) to the terminal acetylene of (XVII) by means of BF3/Et2O in THF gives the 3-butyl-1-ol derivative (XIX), which is condensed with N,O-bis(phenoxy- carbonyl)hydroxylamine (XX) by means of PPh3 and diisopropylazodicarboxylate (DIAD) in THF yielding the final intermediate (XXI). Finally, this compound is treated with ammonia in methanol to obtain the target urea derivative.
WO 0001381 Intermediate (XVI) is treated with BuLi and diisopropylamine in THF giving the chiral acetylenic tetrahydrofuran (XVII). The addition of ethylene oxide (XVIII) to the terminal acetylene of (XVII) by means of BF3/Et2O in THF gives the 3-butyl-1-ol derivative (XIX), which is condensed with N,O-bis(phenoxy- carbonyl)hydroxylamine (XX) by means of PPh3 and diisopropylazodicarboxylate (DIAD) in THF yielding the final intermediate (XXI). Finally, this compound is treated with ammonia in methanol to obtain the target urea derivative.








.gif)
.jpg)
















































































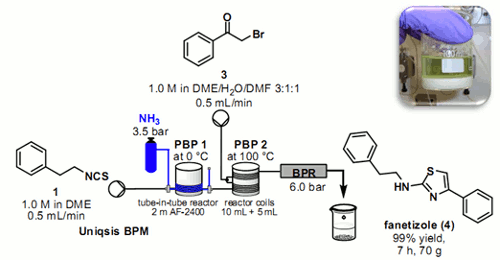























 László Kocsis holds a Masters degree in Bioorganic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2001) and a PhD in Organic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2008). In 2004 he began working as a research chemist at the Reanal Finechemical Company in Budapest, Hungary. He became the Head of the R&D laboratory in 2007 and a manager of production in 2008. In 2011 he joined ThalesNano Inc. as Head of Chemistry. He has experience in organic chemistry, with emphasis on sythesis of amino acid derivatives and peptides, focusing mainly on the following subjects: structure – relationship studies in opiod peptides, methodological studies in the internal solubilization of the sekf-aggregating peptides, industrial scale sythesis of protected amino acid derivatives, and peptides, heterogeneous catalysis, reactions under continuous flow conditions. He is the co-author of 10 pulications and a member of the European Peptide Society.
László Kocsis holds a Masters degree in Bioorganic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2001) and a PhD in Organic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2008). In 2004 he began working as a research chemist at the Reanal Finechemical Company in Budapest, Hungary. He became the Head of the R&D laboratory in 2007 and a manager of production in 2008. In 2011 he joined ThalesNano Inc. as Head of Chemistry. He has experience in organic chemistry, with emphasis on sythesis of amino acid derivatives and peptides, focusing mainly on the following subjects: structure – relationship studies in opiod peptides, methodological studies in the internal solubilization of the sekf-aggregating peptides, industrial scale sythesis of protected amino acid derivatives, and peptides, heterogeneous catalysis, reactions under continuous flow conditions. He is the co-author of 10 pulications and a member of the European Peptide Society.








