 |
|

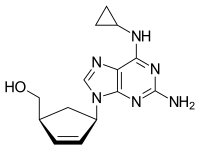
Chemical structure of abacavir
|
{(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopent-2-en-1-yl}methanol
(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
(1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol
Abacavir
Abacavir (ABC) is a powerful nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. [Wikipedia] Chemically, it is a synthetic carbocyclic nucleoside and is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. In vivo, abacavir sulfate dissociates to its free base, abacavir.
Abacavir (ABC)  i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
Viral strains that are resistant to zidovudine (AZT) or lamivudine (3TC) are generally sensitive to abacavir (ABC), whereas some strains that are resistant to AZT and 3TC are not as sensitive to abacavir.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basic health system.[2]
Abacavir is a nucleoside reverse transcriptase inhibitor (NRTI) with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Abacavir is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis. The concentration of drug necessary to effect viral replication by 50 percent (EC50) ranged from 3.7 to 5.8 μM (1 μM = 0.28 mcg/mL) and 0.07 to 1.0 μM against HIV-1IIIB and HIV-1BaL, respectively, and was 0.26 ± 0.18 μM against 8 clinical isolates. Abacavir had synergistic activity in cell culture in combination with the nucleoside reverse transcriptase inhibitor (NRTI) zidovudine, the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine, and the protease inhibitor (PI) amprenavir; and additive activity in combination with the NRTIs didanosine, emtricitabine, lamivudine, stavudine, tenofovir, and zalcitabine.
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| - | J05AF06 | C 14 H 18 N 6 O | 286.34 g / mol | 136470-78-5 |
| succinate | J05AF06 | C 14 H 18 N 6 O · C 4 H 6 O | 356.43 g / mol | 168146-84-7 |
| sulfate | J05AF06 | C 14 H 18 N 6 O · 1 / 2H 2 SO 4 | 670.76 g / mol | 188062-50-2 |
| Systematic (IUPAC) name | |
|---|---|
| {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopent-2-en-1-yl}methanol | |
| Clinical data | |
| Trade names | Ziagen |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699012 |
| Pregnancy cat. | B3 (AU) C (US) |
| Legal status | POM (UK) ℞-only (US) |
| Routes | Oral (solution or tablets) |
| Pharmacokinetic data | |
| Bioavailability | 83% |
| Metabolism | Hepatic |
| Half-life | 1.54 ± 0.63 h |
| Excretion | Renal (1.2% abacavir, 30% 5′-carboxylic acid metabolite, 36% 5′-glucuronide metabolite, 15% unidentified minor metabolites). Fecal (16%) |
| Identifiers | |
| CAS number | 136470-78-5  |
| ATC code | J05AF06 |
| PubChem | CID 441300 |
| DrugBank | DB01048 |
| ChemSpider | 390063 |
| UNII | WR2TIP26VS |
| KEGG | D07057 |
| ChEBI | CHEBI:421707 |
| ChEMBL | CHEMBL1380 |
| NIAID ChemDB | 028596 |
| Chemical data | |
| Formula | C14H18N6O |
| Mol. mass | 286.332 g/mol |
Abacavir is a carbocyclic synthetic nucleoside analogue and an antiviral agent. Intracellularly, abacavir is converted by cellular enzymes to the active metabolite carbovir triphosphate, an analogue of deoxyguanosine-5′-triphosphate (dGTP). Carbovir triphosphate inhibits the activity of HIV-1 reverse transcriptase (RT) both by competing with the natural substrate dGTP and by its incorporation into viral DNA. Viral DNA growth is terminated because the incorporated nucleotide lacks a 3′-OH group, which is needed to form the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation.
Application
-
an antiviral agent, is used in the treatment of AIDS
-
ingibitor convertibility transkriptazы
Classes of substances
-
Adenine (6-aminopurines)
-
Aminoalcohols
-
Cyclopentenes and cyclopentadienes
-
Tsyklopropanы
-
-
-
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2289753 | 2007-01-23 | 2018-05-14 |
| Canada | 1340589 | 1999-06-08 | 2016-06-08 |
| Canada | 2216634 | 2004-07-20 | 2016-03-28 |
| United States | 6641843 | 2000-02-04 | 2020-02-04 |
| United States | 5089500 | 1992-12-26 | 2009-12-26 |
PATENT
US5034394
Synthesis pathway
Abacavir, (-) cis-[4-[2-amino-6-cyclopropylamino)-9H-purin-9-yl]-2-cyclopenten-yl]-1 – methanol, a carbocyclic nucleoside which possesses a 2,3-dehydrocyclopentene ring, is referred to in United States Patent 5,034,394 as a reverse transcriptase inhibitor. Recently, a general synthetic strategy for the preparation of this type of compound and intermediates was reported [Crimmins, et. al., J. Org. Chem., 61 , 4192-4193 (1996) and 65, 8499-8509-4193 (2000)].
-
Abacavir is the International Nonproprietary Name (INN) of {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol and CAS No. 136470-78-5. Abacavir and therapeutically acceptable salts thereof, in particular the hemisulfate salt, are well-known as potent selective inhibitors of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
-
![Figure imgb0001]()
-
EP 434450-A discloses certain 9-substituted-2-aminopurines including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
-
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring.
-
According to the teachings of EP 434450-A , the abacavir base is finally isolated by trituration using acetonitrile (ACN) or by chromatography, and subsequently it can be transformed to a salt of abacavir by reaction with the corresponding acid. Such isolation methods (trituration and chromatography) usually are limited to laboratory scale because they are not appropriate for industrial use. Furthermore, the isolation of the abacavir base by trituration using acetonitrile gives a gummy solid (Example 7) and the isolation by chromatography (eluted from methanol/ethyl acetate) yields a solid foam (Example 19 or 28).
-
Other documents also describe the isolation of abacavir by trituration or chromatography, but always a gummy solid or solid foam is obtained (cf. WO9921861 and EP741710 ), which would be difficult to operate on industrial scale.
-
WO9852949 describes the preparation of abacavir which is isolated from acetone. According to this document the manufacture of the abacavir free base produces an amorphous solid which traps solvents and is, therefore, unsuitable for large scale purification, or for formulation, without additional purification procedures (cf. page 1 of WO 9852949 ). In this document, it is proposed the use of a salt of abacavir, in particular the hemisulfate salt which shows improved physical properties regarding the abacavir base known in the art. Said properties allow the manufacture of the salt on industrial scale, and in particular its use for the preparation of pharmaceutical formulations.
-
However, the preparation of a salt of abacavir involves an extra processing step of preparing the salt, increasing the cost and the time to manufacture the compound. Generally, the abacavir free base is the precursor compound for the preparation of the salt. Thus, depending on the preparation process used for the preparation of the salt, the isolation step of the abacavir free base must also be done.
The structure of abacavir corresponds to formula (I):

………………………………
http://www.google.co.in/patents/US5034394
EXAMPLE 21(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
The title compound of Example 7, (2.00 g, 6.50 mmol) was dissolved in 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (Aldrich, 20 mL). Phosphoryl chloride (2.28 mL, 24.0 mmol) was added to the stirred, cooled (-10° C.) solution. After 3 minutes, cold water (80 mL) was added. The solution was extracted with chloroform (3×80 mL). The aqueous layer was diluted with ethanol (400 mL) and the pH adjusted to 6 with saturated aqueous NaOH. The precipitated inorganic salts were filtered off. The filtrate was further diluted with ethanol to a volume of 1 liter and the pH adjusted to 8 with additional NaOH. The resulting precipitate was filtered and dried to give the 5′-monophosphate of (±)-cis-4-[2-amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol as white powder (4.0 mmoles, 62% quantitated by UV absorbance); HPLC analysis as in Example 17 shows one peak. This racemic 5′ -monophosphate was dissolved in water (200 mL) and snake venom 5′-nucleotidase (EC 3.1.3.5) from Crotalus atrox (5,000 IU, Sigma) was added. After incubation at 37° C. for 10 days, HPLC analysis as in Example 17 showed that 50% of the starting nucleotide had been dephosphorylated to the nucleoside. These were separated on a 5×14 cm column of DEAE Sephadex A25 (Pharmacia) which had been preequilibrated with 50 mM ammonium bicarbonate. Title compound was eluted with 2 liters of 50 mM ammonium bicarbonate. Evaporation of water gave white powder which was dissolved in methanol, adsorbed on silica gel, and applied to a silica gel column. Title compound was eluted with methanol:chloroform/1:9 as a colorless glass. An acetonitrile solution was evaporated to give white solid foam, dried at 0.3 mm Hg over P2 O5 ; 649 mg (72% from racemate); 1 H-NMR in DMSO-d6 and mass spectrum identical with those of the racemate (title compound of Example 7); [α]20 D -48.0°, [α]20 436 -97.1°, [α]20 365 -149° (c=0.14, methanol).
Anal. Calcd. for C15 H20 N6 O.0.10CH3 CN: C, 59.96; H, 6.72; N, 28.06. Found: C, 59.93; H, 6.76; N, 28.03.
Continued elution of the Sephadex column with 2 liters of 100 mM ammonium bicarbonate and then with 2 liters of 200 mM ammonium bicarbonate gave 5′-monophosphate (see Example 22) which was stable to 5′-nucleotidase.
…………………………………………
| Синтез a) |
|---|
        |
| Синтез b) |
     |
| Preparation c) |
    |
| Synthesis d) |
 |

An enantiopure β-lactam with a suitably disposed electron withdrawing group on nitrogen, participated in a π-allylpalladium mediated reaction with 2,6-dichloropurine tetrabutylammonium salt to afford an advanced cis-1,4-substituted cyclopentenoid with both high regio- and stereoselectivity. This advanced intermediate was successfully manipulated to the total synthesis of (−)-Abacavir.

http://pubs.rsc.org/en/content/articlelanding/2012/ob/c2ob06775g#!divAbstract
………………………………….
http://www.google.com.ar/patents/EP2085397A1?cl=en
Example 1: Preparation of crystalline Form I of abacavir base using methanol as solvent
-
[0026]Abacavir (1.00 g, containing about 17% of dichloromethane) was dissolved in refluxing methanol (2.2 mL). The solution was slowly cooled to – 5 °C and, the resulting suspension, was kept at that temperature overnight under gentle stirring. The mixture was filtered off and dried under vacuum (7-10 mbar) at 40 °C for 4 hours to give a white solid (0.55 g, 66% yield, < 5000 ppm of methanol). The PXRD analysis gave the diffractogram shown in FIG. 1.
……………………………………..
http://www.google.com/patents/WO2008037760A1?cl=en
Abacavir, is the International Nonproprietary Name (INN) of {(1 S,4R)-4-[2- amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol, and CAS No. 136470-78-5. Abacavir sulfate is a potent selective inhibitor of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
The structure of abacavir hemisulfate salt corresponds to formula (I):

(I)
EP 434450-A discloses certain 9-substituted-2-aminopuhnes including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring. Pyrimidine compounds which have been identified as being useful as intermediates of said preparation processes include N-2-acylated abacavir intermediates such as N-{6- (cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H-purin- 2-yl}acetamide or N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-
(hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide. The removal of the amino protective group of these compounds using acidic conditions is known in the art. According to Example 28 of EP 434450-A, the amino protective group of the N-{6-(cyclopropylamino)-9-[(1 R,4S)-4- (hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide is removed by stirring with 1 N hydrochloric acid for 2 days at room temperature. The abacavir base, after adjusting the pH to 7.0 and evaporation of the solvent, is finally isolated by trituration and chromatography. Then, it is transformed by reaction with an acid to the corresponding salt of abacavir. The main disadvantages of this method are: (i) the use of a strongly corrosive mineral acid to remove the amino protective group; (ii) the need of a high dilution rate; (iii) a long reaction time to complete the reaction; (iv) the need of isolating the free abacavir; and (v) a complicated chromatographic purification process.
Thus, despite the teaching of this prior art document, the research of new deprotection processes of a N-acylated {(1 S,4R)-4-[2-amino-6- (cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is still an active field, since the industrial exploitation of the known process is difficult, as it has pointed out above. Thus, the provision of a new process for the removal of the amino protective group of a N-acylated {(1 S,4R)-4-[2-amino-6-
(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is desirable.
Example 1 : Preparation of abacavir hemisulfate
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (6.56 g, 18.40 mmol) was slurried in a mixture of isopropanol (32.8 ml) and 10% solution of NaOH (36.1 ml, 92.0 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (32.8 ml) was added. The layers were separated and H2SO4 96% (0.61 ml, 11.03 mmol) was added dropwise to the organic layer. This mixture was cooled to 0-50C and the resulting slurry filtered off.
The solid was dried under vacuum at 40 0C. Abacavir hemisulfate (5.98 g, 97%) was obtained as a white powder.
Example 6: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.0 g, 2.80 mmol) was slurried in a mixture of isopropanol (2 ml) and 10% solution of NaOH (1.1 ml, 2.80 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (2 ml) was added. The aqueous layer was discarded, the organic phase was cooled to 0-5 0C and the resulting slurry filtered off. The solid was dried under vacuum at 400C. Abacavir (0.62 g, 77%) was obtained as a white powder.
Example 7: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetone. Abacavir (0.47 g, 47%) was obtained as a white powder.
Example 8: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetonitrile. Abacavir (0.43 g, 43%) was obtained as a white powder.
Example 9: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in ethyl acetate (150 ml) to afford abacavir (7.2 g, 90%).
Example 10: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in acetone (300 ml) to afford abacavir (7.0 g, 88%).
…………………………………
http://www.google.com/patents/WO2004089952A1?cl=en
Abacavir of formula (1) :

or (1 S,4R)-4-[2-Amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1 – methanol and its salts are nucleoside reverse transcriptase inhibitors. Abacavir sulfate is a nucleoside reverse transcriptase inhibitor and used in the treatment of human immunodeficiency virus infection. Abacavir sulfate and related compounds and their therapeutic uses are disclosed in US 5,034,394.
Crystalline forms of abacavir sulfate have not been reported in the literature. Moreover, the processes described in the literature do not produce abacavir sulfate in a stable, well-defined and reproducible crystalline form. It has now been discovered that abacavir sulfate can be prepared in three stable, well-defined and consistently reproducible crystalline forms.
Example 1
Abacavir free base (3.0 gm, obtained by the process described in example 21 of US 5,034,394) is dissolved in ethyl acetate (15 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 3 hours at 20°C and filtered to give 3.0 gm of form I abacavir sulfate. Example 2 Abacavir free base (3.0 gm) is dissolved in acetone (20 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 6 hours at 25°C and filtered to give 2.8 gm of form I abacavir sulfate.
Example 3 Abacavir free base (3.0 gm) is dissolved in acetonitrile (15 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 2 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 4 Abacavir free base (3.0 gm) is dissolved in methyl tert-butyl ether (25 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 1 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 5 Abacavir free base (3.0 gm) is dissolved in methanol (15 ml) and sulfuric acid (0.3 ml) is added to the solution. The contents then are cooled to 0°C and diisopropyl ether (15 ml) is added. The reaction mass is stirred for 2 hours at about 25°C and the separated solid is filtered to give 3.0 gm of form III abacavir sulfate
…………………………….
http://www.google.com.ar/patents/WO1999021861A1?cl=en
The present invention relates to a new process for the preparation of the chiral nucleoside analogue (1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol (compound of Formula (I)).
The compound of formula (I) is described as having potent activity against human immunodeficiency virus (HIV) and hepatitis B virus (HBV) in EPO34450.

Results presented at the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy (October 4-7, 1994) demonstrate that the compound of formula I has significant activity against HIV comparable to, and if not better than, some current anti HIV drugs, such as zidovudine and didanosine.
Currently the compound of Formula (I) is undergoing clinical investigation to determine its safety and efficacy in humans. Therefore, there exists at the present time a need to supply large quantities of this compound for use in clinical trials.
Current routes of synthesising the compound of formula (I) involve multiple steps and are relatively expensive. It will be noted that the compound has two centres of asymmetry and it is essential that any route produces the compound of formula (I) substantially free of the corresponding enantiomer, preferably the compound of formula (I) is greater than 95% w/w free of the corresponding enantiomer.
Processes proposed for the preparation of the compound of formula (I) generally start from a pyrimidine compound, coupling with a 4-amino-2-cyclopentene-1- methanol analogue, cyciisation to form the imidazole ring and then introduction of the cyclopropylamine group into the 6 position of the purine, such routes include those suggested in EPO434450 and WO9521161. Essentially both routes disclosed in the two prior patent applications involve the following steps:-
(i) coupling (1S, 4R)-4-amino-2-cyclopentene-1 -methanol to N-(4,6-dichloro-5- formamido-2-pyrimidinyl) acetamide or a similar analogue thereof, for example N- (2-amino-4,6-dichloro-5-pyrimidinyl) formamide;
(ii) ring closure of the resultant compound to form the intermediate (1 S, 4R)-4- (2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1 -methanol;
(iii) substituting the halo group by a cyclopropylamino group on the 6 position of the purine ring.
The above routes are multi-step processes. By reducing the number of processing steps significant cost savings can be achieved due to the length of time to manufacture the compound being shortened and the waste streams minimised.
An alternative process suggested in the prior art involves the direct coupling of carbocyclic ribose analogues to the N atom on the 9 position of 2-amino-6-chloro purine. For example WO91/15490 discloses a single step process for the formation of the (1S, 4R)- 4-(2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1- methanol intermediate by reacting (1S, 4R)-4-hydroxy-2-cyclopentene-1 -methanol, in which the allylic hydroxyl group has been activated as an ester or carbonate and the other hydroxyl group has a blocking group attached (for example 1 ,4- bis- methylcarbonate) with 2-amino-6-chloropurine.
However we have found that when synthesising (1S, 4R)-4-(2-amino-6-chloro-9H- purin-9-yl)-2-cyclopentene-1- methanol by this route a significant amount of an N- 7 isomer is formed (i.e. coupling has occurred to the nitrogen at the 7- position of the purine ring) compared to the N-9 isomer desired. Further steps are therefore required to convert the N-7 product to the N-9 product, or alternatively removing the N-7 product, adding significantly to the cost. We have found that by using a transition metal catalysed process for the direct coupling of a compound of formula (II) or (III),

Example 1 (1 S. 4R)-4-[2-Amino-6-(cvclopropylamino)-9H purin-9-vπ-2-cvclopentene-1 – methanol
Triphenylphosphine (14mg) was added, under nitrogen, to a mixture of (1S.4R)- 4-hydroxy-2-cyclopentene -1 -methanol bis(methylcarbonate) (91 mg), 2-amino-6- (cyclopropylamino) purine (90mg), tris(dibenzylideneacetone)dipalladium (12mg) and dry DMF (2ml) and the resulting solution stirred at room temperature for 40 min.
The DMF was removed at 60° in vacuo and the residue partitioned between ethyl acetate (25ml.) and 20% sodium chloride solution (10ml.). The ethyl acetate solution was washed with 20% sodium chloride (2x12ml.) and with saturated sodium chloride solution, then dried (MgSO4) and the solvent removed in vacuo.
The residue was dissolved in methanol (10ml.), potassium carbonate (17mg) added and the mixture stirred under nitrogen for 15h.
The solvent was removed in vacuo and the residue chromatographed on silica gel
(Merck 9385), eluting with dichloromethane-methanol [(95:5) increasing to (90:10)] to give the title compound (53mg) as a cream foam.
δ(DMSO-d6): 7.60 (s.1 H); 7.27 (s,1 H); 6.10 (dt,1 H); 5.86 (dt, 1 H); 5.81 (s,2H); 5.39 (m,1H); 4.75 (t,1H); 3.44 (t,2H); 3.03 (m, 1H): 2.86 (m,1H);2.60 (m,1H); 1.58 (dt, 1 H); 0.65 (m, 2H); 0.57 (m,2H).
TLC SiO2/CHCI3-MeOH (4:1 ) Rf 0.38; det. UN., KMnO4
Trade Names
| Page | Trade name | Manufacturer |
|---|---|---|
| Germany | Kiveksa | GlaxoSmithKline |
| Trizivir | -»- | |
| Ziagen | -»- | |
| France | Kiveksa | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| United Kingdom | Kiveksa | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| Italy | Trizivir | -»- |
| Ziagen | -»- | |
| Japan | Épzikom | -»- |
| Ziagen | -»- | |
| USA | Épzikom | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| Ukraine | Virol | Ranbaksi Laboratories Limited, India |
| Ziagen | GlaksoSmitKlyayn Inc.., Canada | |
| Abamun | Tsipla Ltd, India | |
| Abacavir sulfate | Aurobindo Pharma Limited, India |
Formulations
-
Oral solution 20 mg / ml;
-
Tablets of 300 mg (as the sulfate);
-
Trizivir tablets 300 mg – abacavir in fixed combination with 150 mg of lamivudine and 300 mg zidovudine
ZIAGEN is the brand name for abacavir sulfate, a synthetic carbocyclic nucleoside analogue with inhibitory activity against HIV-1. The chemical name of abacavir sulfate is (1S,cis)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol sulfate (salt) (2:1). Abacavir sulfate is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. It has a molecular formula of (C14H18N6O)2•H2SO4 and a molecular weight of 670.76 daltons. It has the following structural formula:
 |
Abacavir sulfate is a white to off-white solid with a solubility of approximately 77 mg/mL in distilled water at 25°C. It has an octanol/water (pH 7.1 to 7.3) partition coefficient (log P) of approximately 1.20 at 25°C.
ZIAGEN Tablets are for oral administration. Each tablet contains abacavir sulfate equivalent to 300 mg of abacavir as active ingredient and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The tablets are coated with a film that is made of hypromellose, polysorbate 80, synthetic yellow iron oxide, titanium dioxide, and triacetin.
ZIAGEN Oral Solution is for oral administration. Each milliliter (1 mL) of ZIAGEN Oral Solution contains abacavir sulfate equivalent to 20 mg of abacavir (i.e., 20 mg/mL) as active ingredient and the following inactive ingredients: artificial strawberry and banana flavors, citric acid (anhydrous), methylparaben and propylparaben (added as preservatives), propylene glycol, saccharin sodium, sodium citrate (dihydrate), sorbitol solution, and water.
In vivo, abacavir sulfate dissociates to its free base, abacavir. All dosages for ZIAGEN are expressed in terms of abacavir.
History
Abacavir was approved by the Food and Drug Administration (FDA) on December 18, 1998 and is thus the fifteenth approved antiretroviral drug in the United States. Its patent expired in the United States on 2009-12-26.
Links
-
US 5 089 500 (Burroughs Wellcome; 18.2.1992; GB-prior. 27.6.1988).
-
Synthesis a)
-
EP 434 450 (Wellcome Found .; 26.6.1991; appl. 21.12.1990; prior-USA. 22.12.1989).
-
Crimmins, MT et al .: J. Org. Chem. (JOCEAH) 61 4192 (1996).
-
EP 1 857 458 (Solmag; appl. 5.5.2006).
-
EP 424 064 (Enzymatix; appl. 24.4.1991; GB -prior. 16.10.1989).
-
U.S. 6 340 587 (Beecham SMITHKLINE; 22.1.2002; appl. 20.8.1998; GB -prior. 22.8.1997).
-
-
Синтез b)
-
Olivo, HF et al .: J. Chem. Soc., Perkin Trans. 1 (JCPRB4) 1998, 391.
-
-
Preparation c)
-
U.S. 5 034 394 (Wellcome Found .; 23.7.1991; appl. 22.12.1989; GB -prior. 27.6.1988).
-
-
Synthesis d)
-
WO 9 924 431 (Glaxo; appl. 12.11.1998; WO-prior. 12.11.1997).
-
| WO2008037760A1 * | Sep 27, 2007 | Apr 3, 2008 | Esteve Quimica Sa | Process for the preparation of abacavir |
| EP1905772A1 * | Sep 28, 2006 | Apr 2, 2008 | Esteve Quimica, S.A. | Process for the preparation of abacavir |
| US8183370 | Sep 27, 2007 | May 22, 2012 | Esteve Quimica, Sa | Process for the preparation of abacavir |
| EP0434450A2 | 21 Dec 1990 | 26 Jun 1991 | The Wellcome Foundation Limited | Therapeutic nucleosides |
| EP0741710A1 | 3 Feb 1995 | 13 Nov 1996 | The Wellcome Foundation Limited | Chloropyrimide intermediates |
| WO1998052949A1 | 14 May 1998 | 26 Nov 1998 | Glaxo Group Ltd | Carbocyclic nucleoside hemisulfate and its use in treating viral infections |
| WO1999021861A1 | 24 Oct 1997 | 6 May 1999 | Glaxo Group Ltd | Process for preparing a chiral nucleoside analogue |
| WO1999039691A2 * | 4 Feb 1999 | 12 Aug 1999 | Brooks Nikki Thoennes | Pharmaceutical compositions |
| WO2008037760A1 * | 27 Sep 2007 | 3 Apr 2008 | Esteve Quimica Sa | Process for the preparation of abacavir |
References
- Jump up^ SFGate.com
- Jump up^ “WHO Model List of EssentialMedicines”. World Health Organization. October 2013. Retrieved 22 April 2014.
- Jump up^ https://online.epocrates.com/noFrame/showPage.do?method=drugs&MonographId=2043&ActiveSectionId=5
- Jump up^ Mallal, S., Phillips, E., Carosi, G. et al. (2008). “HLA-B*5701 screening for hypersensitivity to abacavir”. New England Journal of Medicine 358: 568–579.doi:10.1056/nejmoa0706135.
- Jump up^ Rauch, A., Nolan, D., Martin, A. et al. (2006). “Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study”. Clinical Infectious Diseases 43: 99–102. doi:10.1086/504874.
- Jump up^ Heatherington et al. (2002). “Genetic variations in HLA-B region and hypersensitivity reactions to abacavir”. Lancet 359: 1121–1122.
- Jump up^ Mallal et al. (2002). “Association between presence of HLA*B5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir”. Lancet359: 727–732. doi:10.1016/s0140-6736(02)07873-x.
- Jump up^ Rotimi, C.N.; Jorde, L.B. (2010). “Ancestry and disease in the age of genomic medicine”. New England Journal of Medicine 363: 1551–1558.
- Jump up^ Phillips, E., Mallal, S. (2009). “Successful translation of pharmacogenetics into the clinic”. Molecular Diagnosis & Therapy 13: 1–9. doi:10.1007/bf03256308.
- Jump up^ Phillips, E., Mallal S. (2007). “Drug hypersensitivity in HIV”. Current Opinion in Allergy and Clinical Immunology 7: 324–330. doi:10.1097/aci.0b013e32825ea68a.
- Jump up^http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm123927.htmAccessed November 29, 2013.
- Jump up^ http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=ca73b519-015a-436d-aa3c-af53492825a1
- Jump up^ Martin MA, Hoffman JM, Freimuth RR et al. (May 2014). “Clinical Pharmacogenetics Implementation Consortium Guidelines for HLA-B Genotype and Abacavir Dosing: 2014 update”. Clin Pharmacol Ther. 95 (5): 499–500. doi:10.1038/clpt.2014.38.PMC 3994233. PMID 24561393.
- Jump up^ Swen JJ, Nijenhuis M, de Boer A et al. (May 2011). “Pharmacogenetics: from bench to byte–an update of guidelines”. Clin Pharmacol Ther. 89 (5): 662–73.doi:10.1038/clpt.2011.34. PMID 21412232.
- Jump up^ Shear, N.H., Milpied, B., Bruynzeel, D.P. et al. (2008). “A review of drug patch testing and implications for HIV clinicians”. AIDS 22: 999–1007.doi:10.1097/qad.0b013e3282f7cb60.
- Jump up^ http://www.drugs.com/fda/abacavir-ongoing-safety-review-possible-increased-risk-heart-attack-12914.html Accessed November 29, 2013.
- Jump up^ Ding X, Andraca-Carrera E, Cooper C et al. (December 2012). “No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis”. J Acquir Immune Defic Syndr. 61 (4): 441–7. doi:10.1097/QAI.0b013e31826f993c.PMID 22932321.
- Illing PT et al. 2012, Nature, doi:10.1038/nature11147
External links
- Full Prescribing Information
- Abacavir pathway on PharmGKB
- Abacavir dosing guidelines from the Clinical Pharmacogenetics Implementation Consortium (CPIC)
- Abacavir dosing guidelines from the Dutch Pharmacogenetics Working Group (DPWG)
EXTRA INFO
How to obtain carbocyclic nucleosides?
Carbocyclic nucleosides are synthetically the most challenging class of nucleosides, requiring multi-step and often elaborate synthetic pathways to introduce the necessary stereochemistry. There are two main strategies for the preparation of carbocyclic nucleosides. In the linear approach a cyclopentylamine is used as starting material and the heterocycle is built in a stepwise manner (see Scheme 1).
Scheme 1: Linear approach for the synthesis of abacavir.[5]
The more flexible strategy is a convergent approach: a functionalized carbocyclic moiety is condensed with a heterocycle rapidly leading to a variety of carbocyclic nucleosides. Initially, we started our syntheses from cyclopentadiene 1 that is deprotonated and alkylated with benzyloxymethyl chloride to give the diene 2. This material is converted by a hydroboration into cyclopentenol 3 or isomerized into two thermodynamically more stable cyclopentadienes 4a,b. With the protection and another hydroboration step to 5 we gain access to an enantiomerically pure precursor for the synthesis of a variety of carbocyclic 2’-deoxynucleosides e.g.:carba-dT, carba-dA or carba-BVDU.[6] The isomeric dienes 4a,b were hydroborated to the racemic carbocyclic moiety 6.

Scheme 2: Convergent approach for the synthesis of carba-dT.
The asymmetric synthesis route and the racemic route above are short and efficient ways to diverse carbocyclic D- or L-nucleosides (Scheme 2). Different heterocycles can be condensed to these precursors leading to carbocyclic purine- and pyrimidine-nucleosides. Beside α- and β-nucleosides, carbocyclic epi- andiso-nucleosides in the 2’-deoxyxylose form were accessable.[7]
What else is possible? The racemic cyclopentenol 6 can be coupled by a modified Mitsunobu-reaction.Moreover, this strategy offers the possibility of synthesizing new carbocyclic nucleosides by functionalizing the double bond before or after introduction of the nucleobase (scheme 3).[8]

Scheme 3: Functionalized carbocyclic nucleosides based on cyclopentenol 6.
Other interesting carbocyclic precursors like cyclopentenol 7 can be used to synthesize several classes of carbocyclic nucleoside analogues, e.g.: 2’,3’-dideoxy-2’,3’-didehydro nucleosides (d4-nucleosides), 2’,3’-dideoxynucleosides (ddNs), ribonucleosides, bicyclic nucleosides or even 2’-fluoro-nucleosides.

Scheme 4: Functionalized carbocyclic thymidine analogues based on cyclopentenol 7.
[1] V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M .C. Nicklaus, R. Agbaria, J. Am. Chem. Soc.2004,126, 543.
[2] A. D. Borthwick, K. Biggadike, Tetrahedron 1992, 48, 571.
[3] H. Bricaud, P. Herdewijn, E. De Clercq, Biochem. Pharmacol. 1983, 3583.
[4] P. L. Boyer, B. C. Vu, Z. Ambrose, J. G. Julias, S. Warnecke, C. Liao, C. Meier, V. E. Marquez, S. H. Hughes, J. Med. Chem. 2009, 52, 5356.
[5] S. M. Daluge, M. T. Martin, B. R. Sickles, D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids 2000,19, 297.
[6] O. R. Ludek, C. Meier, Synthesis 2003, 2101.
[7] O. R. Ludek, T. Kraemer, J. Balzarini, C. Meier, Synthesis 2006, 1313.
[8] M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen, C. Meier, Chem. Eur. J. 2012, 18, 11046-11062.
Filed under: AIDS, Uncategorized Tagged: ABACAVIR, antiretroviral agents, hiv







