Image may be NSFW.
Clik here to view.
Image may be NSFW.
Clik here to view.
Quarfloxin, Itarnafloxin
CAS: 865311-47-3.
Chemical Formula: C35H33FN6O3
Exact Mass: 604.25982
Molecular Weight: 604.67
Elemental Analysis: C, 69.52; H, 5.50; F, 3.14; N, 13.90; O, 7.94
Synonym: CX 3543; CX3543; CX-3543; QuarfloxacinTA1-1B
- CX 3543
- CX-3543
- Itarnafloxin
- Quarfloxacin
- Quarfloxin
- UNII-8M31J5031Q
IUPAC/Chemical name:
5-fluoro-N-(2-((S)-1-methylpyrrolidin-2-yl)ethyl)-3-oxo-6-((R)-3-(pyrazin-2-yl)pyrrolidin-1-yl)-3H-benzo[b]pyrido[3,2,1-kl]phenoxazine-2-carboxamide.
-
5-Fluoro-N-(2-((2S)-1-methylpyrrolidin-2-yl)ethyl)-3-oxo-6-(3-(pyrazin-2- yl)pyrrolidin-1-yl)-3H-benzo(b)pyrido(3,2,1-kl)phenoxazine-2-carboxamide
-
3H-Benzo(b)pyrido(3,2,1-kl)phenoxazine-2-carboxamide, 5-fluoro-N-(2-((2S)- 1-methyl-2-pyrrolidinyl)ethyl)-3-oxo-6-(3-pyrazinyl-1-pyrrolidinyl)-
Quarfloxin, also known as Quarfloxacin and CX-3543, is a fluoroquinolone derivative with antineoplastic activity. Quarfloxin disrupts the interaction between the nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis that is overexpressed in cancer cells; disruption of this G-quadruplex DNA:protein interaction in aberrant rRNA biogenesis may result in the inhibition of ribosome synthesis and tumor cell apoptosis.
CX-3543, developed at Cylene Pharmaceuticals, is a multi-targeting oncogene inhibitor evaluated in phase II clinical studies for the treatment of low or intermediate grade neuroendocrine carcinoma, including carcinoid and islet cell cancer. In 2008, a trial for the treatment of chronic lymphocytic leukemia (CLL) was withdrawn prior to patient enrollment. In 2010, phase I clinical studies for the treatment of solid tumors and for the treatment of lymphoma were terminated upon observation that the modified dose schedule presented no advantage over previously studies schedule solid tumors.
CX-3543 was developed using the company’s Quadruplex Targeting technology which is based on quadruplex motifs in genomic DNA that regulate the expression of clusters of key oncogenes but not normal cellular genes. In 2013, the product was licensed to TetraGene by Cylene Pharmaceuticals on an exclusive, worldwide basis for development for the treatment of cancer. Cylene ceased operations in 2013.
Current developer: Cylene Pharmaceuticals Inc. phase 2
Image may be NSFW.
Clik here to view.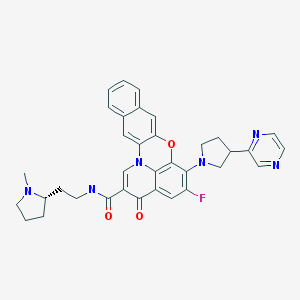
Image may be NSFW.
Clik here to view.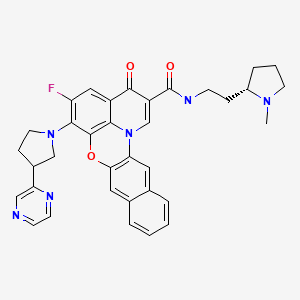
Clinical trial news: Quarfloxin is a ground-breaking small-molecule targeted cancer therapeutic derived from the validated fluoroquinolone class of drugs. Rationally designed to selectively inhibit ribosomal RNA (rRNA) biogenesis in cancer cells, quarfloxin disrupts the interaction between the Nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis and one that is amplified in cancer cells. As a result, quarfloxin selectively induces apoptotic cell death in cancers. Many commercialized cancer therapeutics act indirectly on rRNA Biogenesis through upstream modulators, but quarfloxin is the first agent to directly target this cancer-specific aberrant cell function. According to news released on June 19, 2011, Cylene Pharmaceuticals announced the initiation of a Phase II clinical trial of quarfloxin (CX-3543) in patients with carcinoid/neuroendocrine tumors (C/NET), which are malignant cancers arising from neural crest cells.
Cylene Pharmaceuticals today announced the initiation of a Phase II clinical trial of quarfloxin (CX-3543) in patients with carcinoid/neuroendocrine tumors (C/NET), which are malignant cancers arising from neural crest cells.
“Quarfloxin (CX-3543) is a small molecule that disrupts a protein:rDNA complex that forms in the abnormal nucleoli of cancer cells, thereby selectively inducing apoptotic cell death in cancers,” said Dr. William Rice, President and Chief Executive Officer of Cylene Pharmaceuticals. “Many commercialized cancer therapeutics act on or through the nucleolus, but quarfloxin is the first agent designed to directly target a key function within the nucleolus. Quarfloxin has been well tolerated in humans and has demonstrated signs of biological benefit for patients with C/NET in Phase I clinical trials. Moreover, biodistribution studies revealed that quarfloxin accumulates in the tissues in which C/NET arise.”
In this open-label Phase II trial, quarfloxin will be administered to patients with low or intermediate grade C/NET, including those receiving concomitant treatment with a stable dose of octreotide. This multi-centered study will include an assessment of improvements in patients’ symptoms and biochemical markers, in addition to RECIST tumor response measurements. The first patient was enrolled and treated at Front Range Cancer Specialists in Fort Collins, CO under the care of Robert Marschke Jr., M.D. This study is expected to enroll up to 25 patients at several leading cancer centers.
“The initiation of this Phase II trial with quarfloxin is a major milestone for Cylene, but more importantly, we hope that quarfloxin will be an effective treatment for cancer patients with limited therapeutic alternatives,” added Dr. Daniel Von Hoff, Cylene’s Co-Founder and Vice President, Medical Affairs. “Quarfloxin has demonstrated potent in vivo efficacy against a broad range of tumors and a considerable therapeutic window in preclinical antitumor models, and has a unique profile of concentrating in neural crest tissues. For these reasons, we are enthusiastic about offering a Phase II clinical trial for patients with carcinoid/neuroendocrine tumors.”
About Quarfloxin (CX-3543), a Nucleolus Targeting Agent (NTA)
Quarfloxin is a ground-breaking small-molecule targeted cancer therapeutic derived from the validated fluoroquinolone class of drugs. Rationally designed to selectively inhibit ribosomal RNA (rRNA) biogenesis in cancer cells, quarfloxin disrupts the interaction between the Nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis and one that is amplified in cancer cells. As a result, quarfloxin selectively induces apoptotic cell death in cancers. Many commercialized cancer therapeutics act indirectly on rRNA Biogenesis through upstream modulators, but quarfloxin is the first agent to directly target this cancer-specific aberrant cell function.
About Cylene Pharmaceuticals, Inc.
Cylene Pharmaceuticals is a biotech pharmaceutical company dedicated to the discovery, development and commercialization of targeted small-molecule drugs to treat life-threatening cancers. Cylene has created a diverse portfolio of product candidates, including novel inhibitors of cancer-linked serine/threonine kinases, as well as innovative Nucleolus Targeting Agents (NTAs) that target the abnormal nucleolus functions of cancer cells and selectively kill cancer cells. More information can be found athttp://www.cylenepharma.com.
………………………………………………………..
http://www.google.com/patents/US20060029950
To a series of solutions of the fluoroacid (0.5 mmol) in NMP (3.6 mL) was added the amines NHR1R2 (0.5-2.0 mmol) at room temperature. The vessels were sealed and heated on a 90° C. hotplate with constant stirring for 1-2 hours until the reactions were determined to be complete by HPLC/MS analysis. The reaction mixtures were allowed to cool to room temperature and water was added (20 mL). The resulting precipitates were collected by vacuum filtration and dried under vacuum. In cases where 1.0 equivalent of amine was used, the resulting reaction mixtures were used in the next step “as is.” The resulting solids or solutions were treated with HBTU (2.5 eq.) and DIEA in 3.6 mL NMP and allowed to stir for 30 minutes at room temperature under an inert atmosphere. These solutions were added to a series of amines NHR3R4 (2.5 equivalents) in a 96 well format (Whatman Uniplate, 2 mL) and allowed to react for 2 hours. Methanol was then added (50-100 μL) and the plate was filtered (Whatman Unifilter Polypropylene). The resulting liquids were directly chromatographed on reverse HPLC (Waters Xterra 19×50 mm) with mass directed collection (Micromass ZQ, Waters FCII). The fractions were analyzed for purity (MS TIC, UV) and dried by vacuum evaporation (Savant) with an average yield of 5-10 mg). Examples of substituted quinobenzoxazines analogs are described in Table 1.
Example 48Synthesis of CX-3092 and CX-3543
One method for synthesizing CX-3543 is shown below. As shown in Scheme 2, CX-3543 is synthesized in a convergent manner, assembling the substructures 1, 1A and 2A in the final two synthetic steps (Scheme 2), to form CX-3543 having a 50:50 ratio of RS and SS isomers. CX-3092 may be synthesized in a similar manner using a non-chiral form of 1A.


In more detail, pyrazinopyrrolidine 1A is synthesized via a [3+2] cycloaddition chemistry. Conversion of L-proline 7 to cyano-1-aminopyrrolidine 8 without loss of stereochemistry, followed by reduction provides the chiral 2-aminoethyl-1-methylpyrrolidine 2A in high yield. CX-3543 was found to have a formulated solubility of approximately 20 mg/mL.
Example 70This example describes a method for preparing a substituted benzoxazine analog from reaction of the corresponding ester with an amine, and aluminum chloride.

To a solution of 2,3,4,5-tetrafluorobenzoic acid (100 g, 510 mmol), in methylene chloride (0.5 L) was added oxalyl chloride (68 g, 540 mmol) and DMF (ca 3 drops) and the reaction mixture was allowed to stir at room temperature overnight allowing for the produced gasses to escape. The solvent was removed in vacuo and the vessel was placed on high vacuum (ca 0.5 mm Hg) for 2 hours to afford the acid chloride as a viscous oil (105 g) and was used in the subsequent reaction without further purification.

To a suspension of potassium ethyl malonate (97 g, 570 mmol) and magnesium chloride (55 g, 570 mmol) in acetonitrile and the suspension was chilled to 0° C. To this suspension was added the crude 2,3,4,5-benzoyl chloride (105 g, 520 mmol) over 5 minutes. Triethylamine was slowly added at a rate sufficient to keep the reaction temperature below 10° C. and the mixture was allowed to warm to room temperature and was stirred overnight. The solvent was removed in vacuo and replaced with toluene (300 mL) and 1N HCl (500 mL) was added and the mixture was allowed to stir for 1 hour. The organic layer was separated and washed with 1N HCl (100 mL) and brine (100 mL) and dried over sodium sulfate, filtering over a pad of silica gel (50×100 mm), eluting with ethyl acetate. The solvent was removed in vacuo and the resulting oil was dissolved in ethanol/water (9:1) and was allowed to crystallize overnight. The resulting crystals were Isolated by filtration, washing with ethanol/water (8:2) to afford the ketoester (43.75 g, 166 mmol) as a white crystalline solid.

To a 250 mL round bottom flask was added the tetrafluoroketoester (10.0 g, 37.9 mmol), triethylorthoformate (8.6 mL, 56.8 mmol) and acetic anhydride (7.15 mL, 75.8 mmol) and the reaction mixture was heated to 145° C. for 2 hours. The reaction was allowed to cool to room temperature and placed on high vacuum (ca 0.5 mm Hg) for 1 hour. The resulting oil was dissolved in ethanol (100 mL) and 2-amino-1-naphthol (6.02 g, 37.9 mmol) was added at room temperature and the solution became briefly clear and then product began to precipitate. The reaction was allowed to stir for 2 hours and was then filtered and washed with ethanol (100 mL) to afford the enamine as a yellow solid (12.5 g, 28.9 mmol).

To a solution of the enamine (12.13 g, 27.95 mmol) in dry DMF (50 mL) was added potassium carbonate (4.24 g, 1.1 eq.) and the mixture was heated to 90° C., with constant stirring, for 2 hours. The mixture was allowed to cool to room temperature without stirring and was allowed to remain at room temperature for an additional hour. The crystalline solid was collected by filtration, washing with water. Recrystallization from THF afforded the difluoroester as a white crystalline solid (9.3 g, 23.6 mmol).

To a solution of the difluoroester (1.0 g, 2.5 mmol) in NMP (10 mL) was added N-Boc-3-(2-pyrazino)pyrrolidine (870 mg, 3.5 mmol) and the mixture was heated to reflux for 3 hours. The reaction mixture was then allowed to cool to room temperature and the product was collected by filtration. Crystallization from THF afforded the pyrazine ester as a yellow solid (910 mg, 1.74 mmol).

To a solution of the pyrazine ester (250 mg, 0.48 mmol) and 2-(2-aminoethyl)-1-methylpyrrolidine (80 mg, 0.63 mmol) in methylene chloride at room temperature was added aluminum chloride (83 mg, 0.63 mmol) and the reaction mixture was allowed to stir for 2 hours. The solvent was removed in vacuo and saturated L-tartaric acid was added (5 mL) and the mixture was allowed to stir for 1 hour. Methylene chloride (10 mL) was then added and the mixture was basified with 1N NaOH. The organic layer was separated and washed with a saturated solution of Rochelle’s salt, brine and dried over sodium sulfate. The solvent was removed in vacuo and the resulting solid was dissolved in THF and filtered and the solvent was removed again. The crude solid was recrystallized in ethyl acetate to afford the amide as a yellow solid (225 mg, 0.37 mmol, 98.5% pure).
Example 71
This example describes a method for preparing a substituted benzoxazine analog from reaction of the corresponding carboxylic acid with an amine, and aluminum chloride.

The pyrazinoester (2.0 g, 3.8 mmol) was dissolved in ethanol (100 mL) and conc HCl was added (20 mL) and the mixture was refluxed overnight. The mixture was allowed to cool to room temperature and the solid was collected by vacuum filtration, washing with ethanol to afford the pyrazinoacid as a light tan powder (1.6 g, 3.2 mmol).

To a mixture of the fluoroaminoacid (1.6 g, 3.2 mmol) and HBTU (2.0 g, 5.3 mmol) in NMP (20 mL) was added N,N-diisopropyl-N-ethylamine (1.0 mL, 6 mmol) and the mixture was allowed to stir at room temperature, under argon, for 1 hour (the solution became clear). (S)-2-(2-aminoethyl)-1-methylpyrrolidine (Mizuno, A.; Hamada, Y.; Shioiri, T., Synthesis, 1980, 12 1007)(1.0 mL, 6.9 mmol) was added and the mixture was allowed to stir for 30 minutes. Water (200 mL) was added and the resulting solid was collected by vacuum filtration, washing with water, and dried to afford the pyrazine as a yellow solid. The yellow solid was purified on silica gel (10% MeOH/CH2Cl2 first eluting off impurities followed by eluting with 5% NH4OH/15% MeOH/CH2Cl2. The combined fractions were evaporated to afford the compound as a yellow solid. (1.2 g, 2.0 mmol, 85% pure).
……………………………..
http://www.google.com/patents/WO2007137000A2?cl=en
The present disclosure provides an improved method of treating cancer using a combination of a G-quadruplex-interactive compound that binds to G- quadruplexes in rDNA to release the nucleolin already bound to these G- quadruplexes together with a PARP inhibitor. This results in an increase in apoptosis in cancer cells. The PARP inhibitor can be administered to a patient (human or animal) in need of cancer treatment simultaneously or from 0.1 to 24 hours prior to or 0.1 to 24 after the administration of the G-quadruplex-interactive agent that releases the nucleolin bound to the G-quadruplex and triggers enhanced apoptosis of cancer cells, or the PARP inhibitor and the enhancer of nucleolin binding can be administered simultaneously, with each agent being administered in an amount sufficient to inhibit the growth and/or cell division of cancer (neoplastic) cells, and preferably to cause cancer cell death. In the methods provided herein, the PARP inhibitor can be benzamide (as specifically exemplified) or it can be 3- benzamide, 3-methoxybenzamide, carba-NAD+, nicotinamide, a dihydroisoquinolinone, an isoquinolinone such as 5-methyl-dihydroisoquinolinone, a benzimidazole-4-carboxamide, a 2-aryl-benzimidazole-4-carboxamide, a benzoxazole-4-carboxamide, an N,N-dimethylaminomethyl, pyrrolidinomethyl or bis- benzamide derivative, for example 1 ,5-di(3- carbamoylphenyl)aminocarbonyloxy)pentane, a phthalazinone, a quinazolinone, an isoindolinone, a phenanthhdinone, among others. The G-quadruplex-interactive agent that releases nucleolin from the rDNA bound to the G-quadruplexes and triggers apoptosis of cancer cells is desirably a substituted quinobenzoxazine analog; in an embodiment of the invention, it is CX-3543

(see also US Patent Publication 2006-0029950, which is incorporated by reference herein). This combination chemotherapy can be administered in a single dose, or it can be administered at intervals chosen by a medical or veterinary practitioner.
[0003] The present disclosure further provides compositions comprising a PARP inhibitor and a G-quadruplex-interactive compound that triggers the release of nucleolin from the G-quadruplexes in the rDNA and triggers apoptosis of cancer cells. The compositions desirably further comprises a pharmaceutically acceptable excipient, especially one which is compatible with intravenous administration in human patients. In an embodiment of the invention, the composition comprises benzamide and CX-3543.
…………………………….
WO 2004091504 or http://www.google.com/patents/EP1610759A2?cl=en
|
References: |
1. Bayes, M.; Rabasseda, X.; Prous, J. R., Gateways to clinical trials. Methods Find Exp Clin Pharmacol 2007, 29, (1), 53-71.
2. Brennan, A. B.; Long, C. J.; Bagan, J. W.; Schumacher, J. F.; Spiecker, M. M. Surface topographies for non-toxic bioadhesion control. US20100226943A1.
3. Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C. B.; Proffitt, C.; Trent, K.; Whitten, J. P.; Lim, J. K. C.; Von, H. D.; Anderes, K.; Rice, W. G., Anticancer Activity of CX-3543: A Direct Inhibitor of rRNA Biogenesis. Cancer Res. 2009, 69, (19), 7653-7661.
4. Hurley, L. H.; Guzman, M. Combination cancer chemotherapy. WO2007137000A2, 2007.
5. Lim, J.; Whitten, J. P. Drug administration methods. WO2007143587A1, 2007.
6. Neidle, S., Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 277, (5), 1118-1125.
7. O’Brien, S.; Siddiqui-Jain, A. Targeting quadruplex sequences in human nucleic acids by identifying interacting quinoline and porphyrin derivatives. WO2007056113A2, 2007.
8. Ryckman, D. M.; Drygin, D.; Whitten, J. P.; Anderes, K.; Trent, K.; Darjania, L.; Haddach, M.; O’Brien, S.; Rice, W. G. Methods for treating aberrant cell proliferation disorders. US20080318938A1, 2008.
9. Tian, M.; Zhang, X.; Pan, R.; Zhao, C.; Tang, Y., Structure of G-quadruplex in the oncogene c-myc promoter and small ligands targeting the G-quadruplex. Huaxue Jinzhan 22, (5), 983-992.
10. Whitten, J. P.; O’Brien, S. Methods for treating ophthalmic disorders. US20080318939A1, 2008.
11. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Preparation of fused quinolone analogs which inhibit cell proliferation and/or induce cell apoptosis. US20060074089A1, 2006.
12. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Preparation of fused quinolone analogs which inhibit cell proliferation and/or induce cell apoptosis. WO2006034113A2, 2006.
13. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Process for the preparation of benzothiazole and phenoxazine compounds. US20060063761A1, 2006.
14. Whitten, J. P.; Schwaebe, M.; Siddiqui-Jain, A.; Moran, T. Preparation of substituted quinobenzoxazine analogs as antitumor agents. US20060029950A1, 2006.
| “CX-3543, 386705” ANNUAL DRUG DATA REPORT, PROUS, BARCELONA, ES, vol. 27, no. 4, 2005, page 379, XP009092663 ISSN: 0379-4121 | ||
| 2 | * | CEPEDA, V. ET AL.: “Poly(ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors in Cancer Chemotherapy” RECENT PATENTS ON ANTI-CANCER DRUG DISCOVERY, vol. 1, no. 1, January 2006 (2006-01), pages 39-53, XP007903584 ISSN: 1574-8928 |
| 3 | * | DATABASE INTEGRITY [Online] Prous science; DailyDrugNews.com (Daily Essentials) 22 July 2005 (2005-07-22), “CX-3543 begins phase I cancer trial” XP007903594 retrieved from INTEGRITY.PROUS.COM Database accession no. 386705 |
| 4 | * | JIN ET AL: “In vivo efficacy of CX-3543, a novel c-Myc oncogene inhibitor” PROCEEDINGS OF THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR CANCER RESEARCH, NEW YORK, NY, US, vol. 45, 2004, page ABS. LB-243, XP001537665 ISSN: 0197-016X |
| 5 | * | RICE WILLIAM G ET AL: “Design of CX-3543, a novel multi-targeting antitumor agent” PROCEEDINGS OF THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR CANCER RESEARCH, NEW YORK, NY, US, vol. 46, April 2005 (2005-04), pages 594-ABS. 2530, XP001536592 ISSN: 0197-016X |
| 6 | * | SHIOKAWA D ET AL: “Inhibitors of poly(ADP-ribose) polymerase suppress nuclear fragmentation and apoptotic-body formation during apoptosis in HL-60 cells” FEBS LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 413, no. 1, 11 August 1997 (1997-08-11), pages 99-103, XP004261237 ISSN: 0014-5793 |
| 7 | * | VALERIOTE F ET AL: “SYNERGISTIC INTERACTION OF ANTICANCER AGENTS: A CELLULAR PERSPECTIVE” CANCER CHEMOTHERAPY REPORTS, vol. 59, no. 5, September 1975 (1975-09), pages 895-900, XP009019750 |
| S7910600 | Aug 29, 2008 | Mar 22, 2011 | Cylene Pharmaceuticals, Inc. | Therapeutic kinase modulators |
| US7956064 | Aug 31, 2007 | Jun 7, 2011 | Cylene Pharmaceuticals, Inc. | Fused tricyclic compounds as serine-threonine protein kinase and PARP modulators |
| US8481529 | May 9, 2007 | Jul 9, 2013 | The Arizona Board Of Regents On Behalf Of The University Of Arizona | Combination cancer chemotherapy |
| EP2023935A1 * | Jun 1, 2007 | Feb 18, 2009 | Cylene Pharmaceuticals, Inc. | Drug administration methods |
| WO2007137000A2 * | May 9, 2007 | Nov 29, 2007 | Univ Arizona | Combination cancer chemotherapy |
| WO2007143587A1 * | Jun 1, 2007 | Dec 13, 2007 | Cylene Pharmaceuticals Inc | Drug administration methods |
Filed under: Uncategorized Tagged: CX-3543, Cylene Pharmaceuticals, Itarnafloxin, Quarfloxin, TA1-1B Image may be NSFW.
Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
