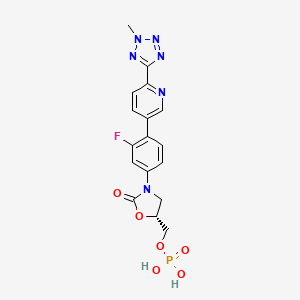
TEDIZOLID PHOSPHATE
[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxo-5-oxazolidinyl]methyl]phosphate,
DA 7157
THERAPEUTIC CLAIM Treatment of complicated skin and skin structure infections
CHEMICAL NAMES
1. 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5- [(phosphonooxy)methyl]-, (5R)-
2. [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxooxazolidin-5- yl]methyl hydrogen phosphate
http://www.ama-assn.org/resources/doc/usan/tedizolid-phosphate.pdf
MOLECULAR FORMULA C17H16FN6O6P
MOLECULAR WEIGHT 450.3
TRADEMARK None as yet
SPONSOR Trius Therapeutics
CODE DESIGNATION TR-701 FA
CAS REGISTRY NUMBER 856867-55-5
Note: This adoption statement supersedes the USAN torezolid phosphate (N09/81), which is hereby rescinded and replaced by the USAN tedizolid phosphate (N10/118).\
……………………………..
Tedizolid, 856866-72-3
(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-5-(hydroxymethyl)-1,3-oxazolidin-2-one
(5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone,
TR 700
- Molecular Formula: C17H15FN6O3
- Average mass: 370.337799
Torezolid (also known as TR-701 and now tedizolid[1]) is an oxazolidinone drug being developed by Trius Therapeutics (originator Dong-A Pharmaceuticals) for complicated skin and skin-structure infections (cSSSI), including those caused by Methicillin-resistantStaphylococcus aureus (MRSA).[2]
As of July 2012, tedizolid had completed one phase III trial, with another one under way. [3]Both trials compare a six-day regimen of tedizolid 200mg once-daily against a ten-day regimen of Zyvox (linezolid) 600mg twice-daily.
The prodrug of tedizolid is called “TR-701″, while the active ingredient is called “TR-700″.[4][5]

Trius Therapeutics will soon be reporting data from its second phase III trial (ESTABLILSH-2) and the recently announced publication of the data from its first phase III trial (ESTABLISH-1) in the Journal of the American Medical Association (JAMA)
- “Trius grows as lead antibiotic moves forward”. 31 Oct 2011.
- “Trius Completes Enrollment In Phase 2 Clinical Trial Evaluating Torezolid (TR-701) In Patients With Complicated Skin And Skin Structure Infections”. Jan 2009.
- http://clinicaltrials.gov/ct2/results?flds=Xf&flds=a&flds=b&term=tedizolid&phase=2&fund=2&show_flds=Y
- PMID 19528279 In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent.
- PMID 19218276 TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens.
…………………………………………………….
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistance. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in the discovery of antibacterial agents.
Several antibacterial agents have been described in the prior art (for example, see PCT International Application Nos. PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178 and PCT/US2009/041200). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.
Various oxazolidinone-containing compounds have been disclosed for use asantibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog
active drug of Formula I is (5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone, i.e.,

These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010).
US Patent Publication No. 20070155798, recently disclosed a series of potently anti-bacterial oxazolidinones including

wherein R═H, PO(OH)2, and PO(ONa)2.
Cubist Announces Submission of New Drug Application for Investigational Antibiotic Tedizolid for Treatment of Serious Skin Infections
LEXINGTON, Mass.–(BUSINESS WIRE)– Cubist Pharmaceuticals, Inc. today announced that it has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of its investigational antibiotic tedizolid phosphate (TR-701). Cubist is seeking approval of tedizolid phosphate for the treatment of acute bacterial skin and skin structure infections (ABSSSI). Tedizolid phosphate is a once daily oxazolidinone being developed for both intravenous (I.V.) and oral administration for the treatment of serious Gram-positive infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
http://www.drugs.com/nda/tedizolid_131023.html
…………………………………………………………..
Espinoza-González NA, Welsh O, de Torres NW, Cavazos-Rocha N, Ocampo-Candiani J, Said-Fernandez S, Lozano-Garza G, Choi SH, Vera-Cabrera L.
Molecules. 2008 Jan 11;13(1):31-40.
…………………………………………..
imp patents
|
12-3-2010
|
OXAZOLIDINONE CONTAINING DIMER COMPOUNDS, COMPOSITIONS AND METHODS TO MAKE AND USE
|
|
|
10-20-2010
|
Oxazolidinone derivatives
|
|
|
7-31-2009
|
NOVEL OXAZOLIDINONE DERIVATIVES
|
…………………………………………..
TEDIZOLID disodium salt
59 nos in
http://www.google.com/patents/US20130102523

 38 nos
38 nos
Tedizolid (formerly known as torezolid or TR-700) is the active hydroxymethyl oxazolidinone having the following formula:

Pharmaceutical prodrugs such as tedizolid phosphate (also referred to as TR-701, torezolid phosphate, and TR-701 “free acid” or FA) have the following formula:

The disodium salt of tedizolid phosphate, has the following structure:

Example 1 Preparation of the Phosphate Monohydrogen Diester, Formula III
In this and the following Examples, “Formula III” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00024
and M=OH.
A 1-L, three-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula Ia below (16.0 g, 0.0499 mol], THF (320 mL, 20 vol) and Et3N (21.9 g, 0.216 mol, 5.0 equiv.).
Figure US20100305069A1-20101202-C00025
POCl3 (3.31 g, 0.0216 mol, 0.5 equiv.) was added dropwise via syringe over 5 minutes. The reaction temperature was maintained below 25° C. The batch was aged for 16 hours at room temperature at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction vessel was then immersed in an ice-water bath and a 500-mL addition funnel charged with 320 mL of H2O was attached to the reaction vessel. When the temperature of the reaction reached 2.7° C., H2O was added drop wise over 30 minutes. The temperature of the reaction was maintained below 10° C. Upon completion of the H2O addition, the ice-water bath was removed and the batch was aged for 3 hours. The solution was transferred to a 2-L round-bottom flask and concentrated under reduced pressure on a rotary evaporator. After removal of most of the THF from the solution, the aqueous mixture was extracted with 5 1-L portions of CH2Cl2:MeOH (9:1). The CH2Cl2 layers were combined and concentrated to a dark oil. This crude material was purified on 200 g of silica gel, eluting with 10% MeOH/CH2Cl2 to 20% 2 N NH3 in MeOH/CH2Cl2. Fractions containing mostly the bis-ester (as judged by TLC Rf=0.3 eluting with 20% 2 N NH3 in MeOH/CH2Cl2) were combined and concentrated under reduced pressure on a rotary evaporator, during which time a white precipitate was observed. The flask containing the slurry was removed from the rotary evaporator and equipped with a magnetic stir bar and allowed to stir while cooling to room temperature over 3 hours, during which time the slurry thickened. The solid was filtered and dried in a vacuum oven at 45° C. for 16 hours to give 3.55 g of bis-ester as an off-white solid (20% yield). HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min. This reaction was repeated and the combined lots of the compound of Formula III (6.7 g) were slurried in 100 mL of MeOH (15 vol). The slurry was heated to 40° C. for 30 minutes and then allowed to cool to room temperature over 1 hour. The off-white solid was filtered and dried in a vacuum oven at 40° C. for 16 hours to give 6.15 g of the compound of Formula III (92% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min.
Example 2 Preparation of the Diphosphate Dihydrogen Diester, Formula IV
In Examples 2-5, “Formula IV” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00026
n=0 and M=O-imidazolium salt.
A 250-mL 3-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula IIa below (5.0 g, 11.1 mmol), carbonyldiimidazole (890 mg, 5.55 mmol, 0.5 equiv.) and DMF (100 mL, 20 vol).
Figure US20100305069A1-20101202-C00027
The suspension was heated to 50° C. and held at that temperature for 4 hours at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction was filtered at 50° C. and dried in a vacuum oven at 50° C. for 24 hours to give 5.15 g of the imidazolium salt (i.e., the compound of Formula IV) as an off-white solid (98% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 94.5% (AUC), tR=14.6 min.
TABLE 1
Method A (Waters XBridge C18 Column)
Time (min) Flow (mL/min) % A % B
0.0 1.0 98.0 2.0
15.0 1.0 5.0 95.0
25.0 1.0 5.0 95.0
27.0 1.0 98.0 2.0
30.0 1.0 98.0 2.0
A = 87% 25 mM ammonium bicarbonate solution in water/13% Acetonitrile
B = Acetonitrile
Wavelength = 300 nm
 disodium salt is TR 701
disodium salt is TR 701
……………………………………
Various oxazolidinone-containing compounds have been disclosed for use as antibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog,
These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010). The latter two applications are assigned to the same assignee as in the present application
………………………………………………………………………………………………………………………..
SYNTHESIS

DESCRIPTION OF COMPDS
10,
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)

………………………………………………………………………………………………………………………….
18
Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
………………………………………………………………………………………………………………………………………………………….
33
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
…………………………………………………………………………………………………………………………………………..
59
(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)

………………………………………………………………………………………………………………………………………………………
72
mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72)

COMPLETE SYNTHESIS
Example 5
Preparation of 2-cyano-5-bromopyridine
In 1 L of dimethylformamide was dissolved 100 g of 2,5-dibromopyridine, 32 g of cupper cyanide and 17.8 g of sodium cyanide were added to the solution at room temperature and the solution was stirred at the temperature of 150° C. for 7 hours for reaction. After being cooled to room temperature, the reaction mixture was added with water and extracted with ethyl acetate. The organic layer was washed with brine, dehydrated, filtered and concentrated in vacuo. The title compound 54 g was obtained. Yield 70%.
1HNMR(CDCl3) δ 8.76(s,1H), 7.98(dd,1H), 7.58(dd,1H)
Example 6
Preparation of 2-(tetrazol-5-yl)-5-bromopyridine
10 g of 2-cyano-5-bromopyridine prepared in the Preparation example 5 was dissolved in 100 ml of dimethylformamide, 5.33 g of sodiumazide, and 4.4 g of ammonium chloride were added to the solution at room temperature, and the solution was stirred at the temperature of 110° C. for 3 hours for reaction. The reaction mixture was added with water and then was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated and concentrated in vacuo thereby to obtain 10.5 g of the title compound. Yield 85%.
Preparation Example 7 Preparation of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 2-(2-methyltetrazol-5-yl)-5-bromopyridine
10.5 g of 2-(tetrazol-5-yl)-5-bromopyridine prepared in the Preparation example 6 was dissolved in 100 ml of dimethylformamide. And then 6.5 g of sodium hydroxide was added to the solution and 9.3 g of iodomethane was slowly added to the solution at the temperature of 0° C. The solution was stirred for 6 hours at room temperature, added with water, extracted with ethyl acetate. And then the organic layer was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to obtain 4 g of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 5 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine.
1) 2-(1-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.77(t,1H), 8.23(dd,1H), 8.04(dd,1H), 4.46(s,3H)
2) 2-(2-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.80(t,1H), 8.13(dd,1H), 7.98(dd,1H), 4.42(s,3H)
Example 1
Preparation of N-Carbobenzyloxy-3-fluoroaniline
3-fluoroaniline 100 g was dissolved in 1 L of tetrahydrofuran (THF) and the solution was added with 150 g (1.8 mol) of sodium bicarbonate (NaHCO3). After being cooled to 0° C., the solution was slowly added with 154 ml of N-carbobenzyloxy chloride (CbzCl) for reaction. While the temperature was maintained at 0° C., the reaction mixture was let to react for 2 hours with stirring. Afterwards, the reaction was extracted with 0.5 L of ethyl acetate. The organic layer, after being separated, was washed with brine, dried over anhydrous magnesium sulfate (MgSO4) and concentrated in vacuo. The residue was washed twice with n-hexane to afford the title compound as white crystal. 132 g. Yield 85%.
Example 2
Preparation of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
132 g of N-carbobenzyloxy-3-fluoroaniline 132 g prepared in the Preparation example 1 was dissolved in 1.3 L of tetrahydrofuran and the solution was cooled to −78° C. 370 ml of n-buthyllitium (n-BuLi, 1.6M/n-hexane) was slowly added to the solution in a nitrogen atmosphere, followed by stirring for 10 min. And 84 ml of (R)-(−)-glycidylbuthylate was slowly added to the reaction mixture, stirred at the same temperature for 2 hours and allowed to react for 24 hours at room temperature. After completion of the reaction, the solution was added with ammonium chloride (HH4Cl) solution and extracted with 0.5 L of ethyl acetate at room temperature. The organic layer, thus separated, was washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was dissolved in 100 ml of ethyl acetate and washed with n-hexane to give white crystals, which were purified to the title compound. 80 g. Yield 70%.
1H NMR (DMSO-d6) δ 7.85(t,1H), 7.58(dd,1H), 7.23(dd,1H), 4.69(m,1H), 4.02 (t,1H), 3.80(dd,1H), 3.60(br dd,2H).
Example 3
Preparation of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 300 ml of acetonitryl was dissolved 30 g of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 2, and 46 g of trifluoroacetic acid silver salt (CF3COOAg) and 43 g of iodide were added to the solution. After being stirred for one day at room temperature, the solution was added with water and was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine and dehydrated. And then the residue was filtered, concentrated in vacuo and dried thereby to form the title compound 44 g. Yield 94%.
1H NMR (DMSO-d6) δ 7.77(t,1H), 7.56(dd,1H), 7.20(dd,1H), 5.20(m,1H), 4.70 (m,1H), 4.07(t,1H), 3.80(m,1H), 3.67(m,2H), 3.56(m,3H)
Example 4
Preparation of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 660 ml of 1,4-dioxan was dissolved 50 g of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 3, 52 g of hexabutylditin ((Bu3Sn)2) and 9.3 g of dichlorobistriphenylphosphinpalladium were added into the solution, and stirred for 2 hours. The solution was filtered using celite and concentrated in vacuo. The residue was purified by column chromatography and 45 g of the title compound was formed.
1H NMR (DMSO-d6) δ 7.74(m,3H), 5.20(t,1H), 4.71(m,1H), 4.08(t,1H), 3.82(dd,1H), 3.68(m,1H), 3.52(m,1H), 1.48(m, 6H), 1.24(m, 6H), 1.06(m,6H), 0.83(t,9H)
COMPD 10
Example 1 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)
In 150 ml of 1-methyl-2-pyrrolidone was dissolved 37 g of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol. The solution was added with 19.7 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine, 10.44 g of lithium chloride and 2.9 g of dichlorobistriphenylphospine palladium(II) at room temperature and then stirred at the temperature of 120° C. for 4 hours. The reaction mixture was added with water and then extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to provide 8 g of the title compound. Yield 26%.
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.70(m,2H), 7.49(dd,1H), 5.25(t,1H), 4.74(m,1H), 4.46(s,3H), 4.14(t,1H), 3.88(dd,1H), 3.68(m,1H), 3.58 (m,1H)
COMPD 18
Example 28 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
In 5 ml of methylenchloride was dissolved 100 mg of the compound 10. The solution was added with 43 mg of diethylaminosulfurtrifloride (DAST) and 0.078 ml of triethylamine and then stirred for 24 hours. After being concentrating, the reaction mixture was purified by column chromatography to obtain the title compound 75 mg. Yield 75%.
1H NMR (DMSO-d6) δ 8.91(s,1H), 8.19(m,2H), 7.74(t,1H), 7.66(dd,1H) 7.49 (dd,1H), 5.06(m,1H), 4.89(m,2H), 4.46(s,3H), 4.23(t,1H), 3.95(dd,1H)
COMPD 33
Example 37 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
In 10 ml of methanol was dissolved 400 mg of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methansulfonyloxymethyl oxazolidin-2-on prepared in the secondary step of the Example 24. The solution was added with 90 mg of sodium methoxide at room temperature and then stirred for one day at room temperature. The solution was extracted with ethyl acetate and the organic layer, thus separated, was washed with water and brine. The organic layer was dehydrated, filtered, concentrated in vacuo and purified by column chromatography to provide the title compound 200 mg. Yield 58%.
1H NMR(CDCl3) δ 8.90(s,1H), 8.29(d,1H), 8.04(d,1H), 7.61(dd,1H), 7.58 (t,1H), 7.38(dd,1H), 4.80(m,1H), 4.45(s,3H), 4.08(t,1H), 3.96(dd,1H), 3.67 (m,2H), 3.43(s,3H)
COMPD 59
Example 58 Preparation of mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) and (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)
1. The Primary Step
In 10 ml of mixture solvent (tetrahydrofuran:methylenchloride=1:1) was dissolved 1 g of compound 10. The solution was added with 0.6 g of tetrazole and 2.3 g of di-tetrabutyl diisoprophylphosphoamidite and stirred for 15 hours at room temperature. The reaction mixture was refrigerated to −78° C., added with 0.7 g of metachloroperbenzoic acid and stirred for 2 hours. After being cooling to −78° C., the reaction mixture was added with metachloroperbenzoic acid (0.7 g). When the reaction mixture was stirred for 2 hours, the temperature of the reaction mixture was raised to room temperature. The reaction mixture was then added with ethyl acetate. The organic layer, thus separated, was washed with sodium bisulfate, sodium bicarbonate and brine, dehydrated, filtered and concentrated in vacuo, followed by purification with column chromatography thereby to provide (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl phosphoric acid ditetrabuthylester (0.71 g, 71%).
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.74(t,1H), 7.68 (dd,1H), 7.49(dd,1H), 4.98(m,1H), 4.46(s,3H), 4.23(t,1H), 4.18(m,1H), 4.09(m,1H), 3.89 (dd,1H), 1.39(s,9H), 1.38(s,9H)
The crystal prepared the above method was dissolved in a mixture of methanol and chloroform. And then the solution added with 3.4 ml of sodium methoxide (0.3M methanol solution) at the room temperature and stirred for 10 hours. The reaction mixture was concentrated to prepare the residue. The residue was crystallized and filtered thereby to obtain the title compound (compound 59) 300 mg.
1H NMR (D2O) δ 8.27(s,1H), 7.56(dd,2H), 7.06(m,2H), 6.90(m,1H), 4.79 (m,1H), 4.63(s,3H), 3.90(m,4H)
COMPD 72
The Secondary Step
In 30 ml of methylenchloride was dissolved the compound (0.7 g) in the Primary Step. The solution was added with 15 ml of trifluoroacetic acid and then stirred for 1 hour at room temperature. The reaction mixture was concentrated in vacuo to prepare the residue. The residue was crystallized with ethanol and ethyl ether to obtain mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) 400 mg.
1H NMR (DMSO-d6) δ 8.92(s,1H), 8.20(m,2H), 7.74(t,1H), 7.66(dd,1H), 7.500(dd,1H), 4.95 (m,1H), 4.46(s,3H), 4.21(t,1H), 4.05(m,2H), 3.91(dd,1H)
………………………………………………………
IMPURITIES
| Organic Impurities in TR-701 FA Drug Substance | |
| Impurity | |
| ‘Name’ | Structure and Chemical Name |
| Rx600013 ‘Des-methyl TR- 701’ |
 |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(2H-1,2,3,4-tetrazol-5- | |
| yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5-yl)methyl | |
| phosphate | |
| Rx600024 ‘Pyrophosphate’ |
 |
| trihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl pyrophosphate | |
| Rx600014 ‘Ring opened’ |
 |
| dihydrogen 3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetraazol-5- | |
| yl)-3-pyridinyl]aniline}-2-hydroxypropyl phosphate | |
| Rx600023 ‘Me-isomer’ |
 |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl phosphate | |
| Rx600025 ‘Overalkylated- phosphorylated impurity’ |
  |
| (R)-1-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-3- | |
| hydroxypropan-2-yl dihydrogen phosphate; | |
| (R)-3-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-2- | |
| hydroxypropyl dihydrogen phosphate | |
| Rx600020 ‘Dimer impurity’ |
 |
| dihydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl- | |
| 2H-1,2,3,4-tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3- | |
| oxazolidin-5-yl]methyl pyrophosphate | |
| Rx600026 “Chloro” |
 |
| (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H- | |
| tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one | |
| Rx600001 TR-700 |
 |
| 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)- | |
| pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one | |
| Rx600022 ‘Bis phosphate’ |
 |
| hydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4- | |
| tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl | |
| phosphate | |
| Rx600042 |
 |
| 3-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-2-hydroxypropyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
| Rx600043 |
 |
| 2-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-1-hydroxyethyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
……………………………………………..
| US4128654 | 10 Feb 1978 | 5 Dec 1978 | E. I. Du Pont De Nemours And Company | 5-Halomethyl-3-phenyl-2-oxazolidinones |
| US4250318 | 9 Aug 1978 | 10 Feb 1981 | Delalande S.A. | Novel 5-hydroxymethyl oxazolidinones, the method of preparing them and their application in therapeutics |
| US4340606 | 23 Oct 1980 | 20 Jul 1982 | E. I. Du Pont De Nemours And Company | 3-(p-Alkylsulfonylphenyl)oxazolidinone derivatives as antibacterial agents |
| US4461773 | 5 Jan 1984 | 24 Jul 1984 | E. I. Dupont De Nemours And Company | P-Oxooxazolidinylbenzene compounds as antibacterial agents |
| US4476136 | 24 Feb 1982 | 9 Oct 1984 | Delalande S.A. | Aminomethyl-5 oxazolidinic derivatives and therapeutic use thereof |
| US4948801 | 29 Jul 1988 | 14 Aug 1990 | E. I. Du Pont De Nemours And Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| US5523403 | 22 May 1995 | 4 Jun 1996 | The Upjohn Company | Tropone-substituted phenyloxazolidinone antibacterial agents |
| US5565571 | 28 Apr 1994 | 15 Oct 1996 | The Upjohn Company | Substituted aryl- and heteroaryl-phenyloxazolidinones |
| US5652238 | 27 Sep 1994 | 29 Jul 1997 | Pharmacia & Upjohn Company | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| US5688792 | 16 Aug 1994 | 18 Nov 1997 | Pharmacia & Upjohn Company | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| US6365751 | 17 Apr 2001 | 2 Apr 2002 | Zeneca Ltd. | Antibiotic oxazolidinone derivatives |
| US6627646 * | 17 Jul 2001 | 30 Sep 2003 | Sepracor Inc. | Norastemizole polymorphs |
| US6689779 | 18 May 2001 | 10 Feb 2004 | Dong A Pharm. Co., Ltd. | Oxazolidinone derivatives and a process for the preparation thereof |
| US7129259 | 1 Dec 2004 | 31 Oct 2006 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US7141583 | 23 Apr 2001 | 28 Nov 2006 | Astrazeneca Ab | Oxazolidinone derivatives with antibiotic activity |
| US7144911 | 24 Dec 2003 | 5 Dec 2006 | Deciphera Pharmaceuticals Llc | Anti-inflammatory medicaments |
| US7202257 | 6 Jul 2004 | 10 Apr 2007 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US7396847 | 9 Sep 2002 | 8 Jul 2008 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7462633 | 29 Jun 2004 | 9 Dec 2008 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US7473699 | 25 Feb 2003 | 6 Jan 2009 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US7498350 | 24 Nov 2003 | 3 Mar 2009 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US7816379 | 17 Dec 2004 | 19 Oct 2010 | Dong-A Pharm. Co., Ltd. | Oxazolidinone derivatives |
| US20020115669 | 29 Aug 2001 | 22 Aug 2002 | Wiedeman Paul E. | Oxazolidinone chemotherapeutic agents |
| US20030166620 | 18 May 2001 | 4 Sep 2003 | Jae-Gul Lee | Novel oxazolidinone derivatives and a process for the preparation thereof |
| US20040180906 | 24 Dec 2003 | 16 Sep 2004 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20050038092 | 29 Jun 2004 | 17 Feb 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20050107435 | 9 Sep 2002 | 19 May 2005 | Gravestock Michael B. | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US20050288286 | 6 Jul 2004 | 29 Dec 2005 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20060116386 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US20060116400 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline derivatives as antibacterial agents |
| US20060270637 | 24 Feb 2004 | 30 Nov 2006 | Astrazeneca Ab | Hydroxymethyl substituted dihydroisoxazole derivatives useful as antibiotic agents |
| US20070155798 | 17 Dec 2004 | 5 Jul 2007 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20070185132 | 29 Jun 2004 | 9 Aug 2007 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereo |
| US20070191336 | 23 Dec 2004 | 16 Aug 2007 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070203187 | 22 Jan 2007 | 30 Aug 2007 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20070208062 | 24 May 2005 | 6 Sep 2007 | Astrazeneca Ab | 3-(4-(2-dihydroisoxazol-3-ylpyridin-5-yl)phenyl)-5-triazol-1-ylmethyloxazolidin-2-one derivatives as mao inhibitors for the treatment of bacterial infections |
| US20080021012 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-[4-{6-Substituted Alkanoyl Pyridin-3-Yl}-3-Phenyl]-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones As Antibacterial Agents |
| US20080021071 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-{4-(Pyridin-3-Yl) Phenyl}-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20080064689 | 24 May 2004 | 13 Mar 2008 | Astrazeneca Ab | 3-[4-(6-Pyridin-3-Yl)-3-Phenyl] -5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20090018123 | 19 Jun 2006 | 15 Jan 2009 | Milind D Sindkhedkar | Oxazolidinones Bearing Antimicrobial Activity Composition and Methods of Preparation |
| US20090192197 | 16 Sep 2008 | 30 Jul 2009 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20100093669 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| US20100227839 | 3 Feb 2010 | 9 Sep 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin- 5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| AU2004299413A1 | Title not available | |||
| AU2009200606A1 | Title not available | |||
| CA2549062A1 | 17 Dec 2004 | 30 Jun 2005 | Chong Hwan Cho | Novel oxazolidinone derivatives |
| CN101982468A | 17 Dec 2004 | 2 Mar 2011 | 东亚制药株式会社 | Novel oxazolidinone derivatives and pharmaceutical compositions comprising the derivatives |
| EP0312000A1 | 12 Oct 1988 | 19 Apr 1989 | The Du Pont Merck Pharmaceutical Company | Aminomethyl oxooxazolidinyl aroylbenzene derivatives useful as antibacterial agents |
| EP0352781A2 | 27 Jul 1989 | 31 Jan 1990 | The Du Pont Merck Pharmaceutical Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| EP1699784A1 | 17 Dec 2004 | 13 Sep 2006 | Dong-A Pharmaceutical Co., Ltd. | Novel oxazolidinone derivatives |
| EP2305657A2 | 17 Dec 2004 | 6 Apr 2011 | Dong-A Pharmaceutical Co., Ltd. | Oxazolidinone derivatives |
| EP2435051A1 | 27 May 2010 | 4 Apr 2012 | Trius Therapeutics | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
| IN236862A1 | Title not available | |||
| JPS5799576A | Title not available | |||
| KR20110071107A | Title not available | |||
| NZ547928A | Title not available | |||
| NZ575842A | Title not available | |||
| WO1993009103A1 | 5 Oct 1992 | 13 May 1993 | Upjohn Co | Substituted aryl- and heteroarylphenyloxazolidinones useful as antibacterial agents |
| WO1993023384A1 | 21 Apr 1993 | 25 Nov 1993 | Michael Robert Barbachyn | Oxazolidinones containing a substituted diazine moiety and their use as antimicrobials |
| WO1995007271A1 | 16 Aug 1994 | 16 Mar 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO1995014684A1 | 27 Sep 1994 | 1 Jun 1995 | Michel R Barbachyn | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| WO2001094342A1 | 18 May 2001 | 13 Dec 2001 | Cho Jong Hwan | Novel oxazolidinone derivatives and a process for the preparation thereof |
| WO2002081470A1 | 3 Apr 2002 | 17 Oct 2002 | Astrazeneca Ab | Oxazolidinones containing a sulfonimid group as antibiotics |
| WO2003022824A1 | 9 Sep 2002 | 20 Mar 2003 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| WO2003035648A1 | 23 Oct 2002 | 1 May 2003 | Astrazeneca Ab | Aryl substituted oxazolidinones with antibacterial activity |
| WO2003047358A1 | 2 Dec 2002 | 12 Jun 2003 | Vaughan Leslie Crow | Cheese flavour ingredient and method of its production |
| WO2003072575A1 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring) methyl-oxazolidinone derivatives and their use as antibacterial agents |
| WO2003072576A2 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | Oxazolidinone derivatives, processes for their preparation, and pharmaceutical compositions containing them |
| WO2004048350A2 | 24 Nov 2003 | 10 Jun 2004 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| WO2004083205A1 | 16 Mar 2004 | 30 Sep 2004 | Astrazeneca Ab | Antibacterial 1, 3- oxazolidin -2- one derivatives |
| WO2005005398A2 | 29 Jun 2004 | 20 Jan 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| WO2005051933A1 | 23 Nov 2004 | 9 Jun 2005 | Vijay Kumar Kaul | An improved process for the synthesis of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-1-carboxylic acid tert-butyl ester, a key intermediate for oxazolidinone antimicrobials and compounds prepared thereby |
| WO2005058886A1 | 17 Dec 2004 | 30 Jun 2005 | Dong A Pharm Co Ltd | Novel oxazolidinone derivatives |
| WO2005116017A1 | 24 May 2005 | 8 Dec 2005 | Astrazeneca Ab | Process for the preparation of aryl substituted oxazolidinones as intermediates for antibacterial agents |
| WO2006038100A1 | 6 Oct 2005 | 13 Apr 2006 | Ranbaxy Lab Ltd | Oxazolidinone derivatives as antimicrobials |
| WO2007023507A2 | 19 Jun 2006 | 1 Mar 2007 | Milind D Sindkhedkar | Oxazolidinones bearing antimicrobial activity composition and methods of preparation |
| WO2007138381A2 | 13 Oct 2006 | 6 Dec 2007 | Delorme Daniel | Phosphonated oxazolidinones and uses thereof for the prevention and treatment of bone and joint infections |
| WO2010042887A2 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| WO2010091131A1 | 3 Feb 2010 | 12 Aug 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| WO2010138649A1 | 27 May 2010 | 2 Dec 2010 | Trius Therapeutics, Inc. | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
……………………………………………………………………………………….


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007
Filed under: Uncategorized Tagged: anthony crasto, medicinal chemistry, organic chemistry, organic synthesis, PROCESS, Tedizolid, torezolid, world drug tracker







