![GSK744.svg]()
Cabotegravir, GSK 744,
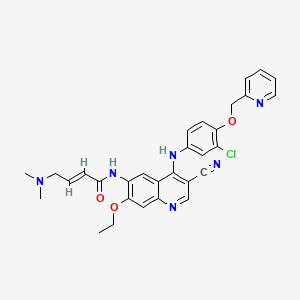
(3S,11aR)-N-(2,4-Difluorobenzyl)-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide
OTHER ISOMER
(3R,11 aS)-N-[(2,4-Diflυorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
VIIV HEALTHCARE …INNOVATOR
![2D chemical structure of 1051375-10-0]()
GSK744 (also known as S/GSK1265744) is an investigational new drug under development for the treatment of HIV infection. It is anintegrase inhibitor, with a carbamoyl pyridone structure similar to dolutegravir. In investigational studies, the agent has been packaged into nanoparticles (GSK744LAP) conferring an exceptionally long half-life of 21–50 days following a single dose. In theory, this would make possible suppression of HIV with dosing as infrequently as once every three months.[1]
S-265744 LAP is in phase II clinical development at Shionogi-GlaxoSmithKline for the treatment of HIV infection. Phase III clinical trials had been ongoing for this indication; however, no recent development has been reported for this study.
![]()
The human immunodeficiency virus (“HIV”) is the causative agent for acquired immunodeficiency syndrome (“AIDS”), a disease characterized by the destruction of the immune system, particularly of CD4+ T-cells, with attendant susceptibility to opportunistic infections, and its precursor Al DS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. HIV is a retrovirus; the conversion of its RNA to DNA is accomplished through the action of the enzyme reverse transcriptase. Compounds that inhibit the function of reverse transcriptase inhibit replication of HIV in infected cells. Such compounds are useful in the prevention or treatment of HIV infection in humans.
A required step in HIV replication in human T-cells is the insertion by virally-encoded integrase of proviral DNA into the host cell genome. Integration is believed to be mediated by integrase in a process involving assembly of a stable nucleoprotein complex with viral DNA sequences, cleavage of two nucleotides from the 3′ termini of the linear proviral DNA and covalent joining of the recessed 3′ OH termini of the proviral DNA at a staggered cut made at the host target site. The repair synthesis of the resultant gap may be accomplished by cellular enzymes. There is continued need to find new therapeutic agents to treat human diseases. HIV integrase is an attractive target for the discovery of new therapeutics due to its important role in viral infections, particularly HIV infections. Integrase inhibitors are disclosed in WO2006/116724.
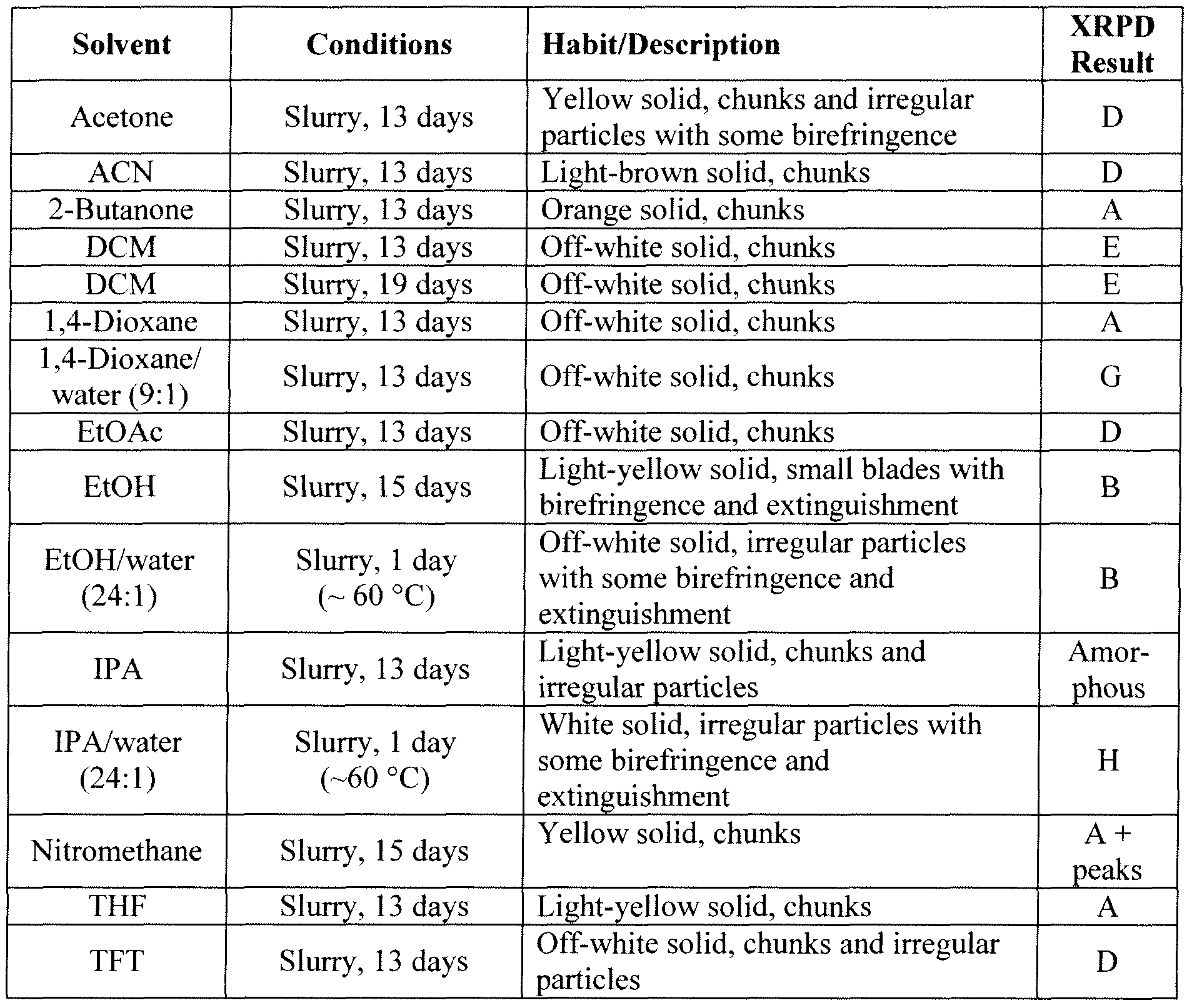
(3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, a compound of formula (I), also referred to as compound (I), has proven antiviral activity against human immunodeficiency virus (HIV).
The present invention features pharmaceutical compositions comprising the active ingredient (3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, or a pharmaceutically acceptable salt thereof, suitable for administration once monthly or longer.
Methods for the preparation of a compound of formula (I) are described in WO 2006/1 16764, WO2010/01 1814, WO2010/068262, and WO2010/068253
WO 2006116764
http://www.google.com/patents/WO2006116764A1?cl=en
![Figure imgf000058_0001]()
[Chemical formula 68] is UNDESIRED ISOMER………..amcrasto@gmail.com
![Figure imgf000122_0001]()
Example Z-1:
(3R,11 aS)-N-[(2,4-Diflυorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a
-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt.
(3R,11aS)-N-[(2,4-Diflυorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,
3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0,14 mL, 1.74 mmol) and 10 drops of glacial acetic acid.
The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added
to the mixture and the solvents removed in vacuo and the material was purified via
silica gel chromatography (2% CH3OH/CH2CI2 gradient elution) to give
(3R),11aS)-N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,
3,5,7, 1 l , 11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazinc-8-carboxamide (396
mg, 92%) as a glass, JH NMR (CDCIo) δ 10.38 (m, 1 H), 8.42 (s, 1 H), 7,54-7,53 (m, 2
H), 7,37-7.24 (m, 4 H), 6.83-6,76 (m, 2 H), 5.40 (d, J = 10.0 Hz, 1 H), 5.22 (d, J = 10,0
Hz, 1 H), 5.16 (dd, J – 9,6, 6.0 Hz, 1 H), 4,62 (m, 2 H), 4.41 (m, 1 H), 4.33-4.30 (m, 2
H), 3.84 (dd, J= 12.0, 10.0 Hz, 1 H), 3.63 (dd, J= 8,4, 7.2 Hz, 1 H), 1.37 (d, J= 6.0 Hz,
3 H); ES+ MS: 496 (M+1).
b)
(3R, 11aS)-N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 1la
-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8vcarboxamide sodium salt. To a
solution of
(37?, 11aS)-N-[(2,4-difluo]-ophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy] -2,
3,5,7,11,11 a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396
mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was
bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture
was filtered through Celite with methanol and dichloromethanc. The filtrate was
concentrated in vacuo to give
(3R, l] aS)-N-f(2,4-difliιorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, υ , 11a- hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide as a pink tinted
white solid (278 mg, 86%), 1H NMR (ODCU) δ 11.47 (m, 1 H), 10.29 (m, 1 H), 8,32 (s,
1 H), 7.36 (m, 1 H), 6.82 (m, 2 H), 5.31 (dd, J – 9.6, 3.6 Hz, 1 H), 4.65 (m, 2 H),
4,47-4,38 (m, 3 H), 3.93 (dd, J= 12.0, 10.0 Hz, 1 H), 3,75 (m, 1 H), 1.49 (d, J= 5.6 Hz,
3 H); BS1 MS: 406 (M+ 1). The above material (278 mg, 0,66 mmol) was taken up
m cthanol (10 mL) and treated with 1 Nsodium hydroxide (aq) (0.66 mL, 0.66 mmol).
The resulting suspension was stirred at room temperature for 30 min, Ether was
added and the liquids were collected to provide the sodium salt of the title compound
as a white powder (291 mg, 99%).‘ 1H NMR (OMSO- do) δ 30.68 (m, 1 H), 7,90 (s, 1 H),
7.35 (m, 1 H), 7.20 (m, 1 H), 7,01 (m, 1 H), 5,20 (m, 1 H), 4,58 (m, I H), 4.49 (m, 2 H),
4.22 (m, 2 H), 3 74 (dd, J= 11.2, 10.4 Hz, 1 H), 3.58 (m, 1 H), 1.25 (d, J=- 4.4 Hz, 3 H)
DESIRED ISOMER………… ANY ERROR ………….amcrasto@gmail.com
Example Z-9-
(3£ 11aΛ^N-[(2.4-D-fluoroDhonyl)methyl] -6-hvdroxy-3-methyl-5.7-dioxo-2,3,5.7, n , 11 a
-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazino-8-carboxamide sodium salt.
The title compound was made in two steps using a similar process to that described
in example Z-I. 16a (510 mg, 1.08 mmol) and (2«5)-2-amino-1-propanol (0.17 mL, 2,17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give
(3S, 11aR)-i\A[(2,4-diflιιorophenyl)methyl]-3-methyl-5,7-d.ioxo-6-[(phenylmethyl)oxy]-2,
3,5,7,11,1la-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500
mg, 93%). This material was hydrogenated in a second step as described in example
Z- I to give
3S, 11a R)-7N-[(2,4-Diiαuorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a-
hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyraziine-8-carboxamide (386 mg, 94%) as a
tinted white solid. Η NMR (CDCL3) δ 11.46 (m, 1 H), 10.28 (m, 1 H), 8.32 (s, 1 H),
7.35 (m, 1 H), 6.80 (m, 2 H), 5.30 (dd, J = 10.0, 4.0 Hz, 1 H), 4.63 (m, 2 H), 4.48-4.37
(m, 3 H), 3.91 (dd, J = 12.0, 10.0 Hz, 1 H), 3.73 (m, 1 H), 1.48 (d, J – 6.0 Hz, 3 H);
ES 1 MS: 406 (M+ 1). This material (385 mg, 0.95 mmol) was treated with sodium
hydroxide (0,95 mL, 1.0 M, 0.95 mmol) m ethanol (15 mL) as described in example Z-1
to provide its corresponding sodrum sail (381 mg, 94%) as a white solid. 1H NMR
(DMSO- Λ) δ 10.66 (m, 1 PI), 7.93 (s, 1 H), 7.33 (m, 1 H), 7.20 (m, 1 H), 7.01 (m, 1 H),
5.19 (m, 1 H), 4.59 (m, 1 H), 4 48 (m, 2 H), 4.22 (m, 2 H), 3,75 (m, 1 H), 3.57 (m, 1 H),
1.24 (d, J= 5 6 Hz, 3 H).
…………………..
WO 2010068253
http://www.google.com/patents/WO2010068253A1?cl=en
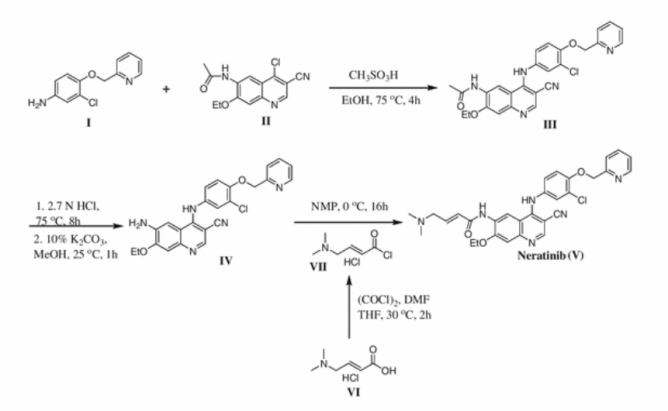
Example A
The starting material of Example A is compound 8, which is identical to formula (Ia). Thus, Example A depicts a process in providing an intermediate for the compound of formula 17 below which is isomeric to the compound ZZ-2 at page 237 of WO 2006/116764 to Brian Johns et al.
14
Example Aa After dissolution of mixture of 320 g of compound 8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 mL of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 °C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 mL of cold 2-propanol and drying provided 167 g of compound 14 (52% yield) as a crystal. 1H NMR(300 MHz1 CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1H), 5.46 (d, J = 10.5 Hz, 1H), 5.23 (d, J = 10.2 Hz, 1H), 5.20 (dd, J = 3.9, 9.6 Hz, 1H), 4.46- 4.34 (m, 1H)1 4.31 (dd, J = 6.6, 8.7 Hz, 1H)1 4.14 (dd, J = 3.9, 12.3 Hz1 1H)1 3.79 (dd, J = 9.9, 12.3 Hz1 1 H), 3.62 (dd, J = 6.9, 8.7 Hz1 1 H), 1.38 (d, J = 6.3 Hz1 3H).
Example Ab
To slurry of 156 g of compound 14 (1.0 eq.) in 780 ml_ of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound 15 (84% yield) as a crystal.
1H NMR(300 MHz, DMSO-CT6) δ 8.37 (s, 1H), 7.55-7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1H), 5.18 (d, J = 10.8 Hz, 1H), 5.03 (d, J = 10.5 Hz, 1H), 4.53 (dd, J = 3.6, 12.0 Hz, 1H)1 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 11.7 Hz1 1H), 3.64 (dd, J = 5.7, 8.1 Hz1 1 H)1 1.27 (d, J = 6.3 Hz1 3H).
Example Ac
Under carbon mono-oxide atmosphere, a mixture of 163 g of compound 15 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 ml_ of 2,4-difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 11.3 g of
Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O1 the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt. The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound 16 (96% yield) as foam.
1H NMR(300 MHz, CDCI3) δ 10.38 (t, J = 6.3 Hz1 1H)1 8.39 (s, 1H)1 7.75-7.25 (m, 7H), 6.90-6.70 (m, 2H), 5.43 (d, J = 10.2 Hz, 1H), 5.24 (d, J = 10.2 Hz, 1H)1 5.19 (dd, J = 3.9, 9.9 Hz, 1H)1 4.63 (d, J = 6.0 Hz, 2H), 4.50-4.25 (m, 3H)1 3.86 (dd, J = 9.9, 12.3 Hz, 1H), , 3.66 (dd, J = 6.9, 8.4 Hz1 1 H), 1.39 (d, J = 6.0 Hz, 3H).
Example Ad
Under hydrogen atmosphere, a mixture of 184 g of compound 16 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1), the filtrate was concentrated. After 200 ml_ of AcOEt was added to the residue, filtration afforded crude solid of compound 17. The precipitates were combined and extracted with 4.0 L of CHCl3/MeOH(5/1). After concentration of the CHCI3ZMeOH solution and addition of 250 ml_ of AcOEt to the residue, filtration afforded crude solid of compound 17. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated (three times). After cooling of the residue, filtration and drying provided 132 g of compound 17 (88% yield) as a crystal. 1H NMR(300 MHz, DMSO-cfe) δ 11.47 (brs, 1H), 10.31 (t, J = 6.0 Hz, 1H), 8.46 (s, 1H), 7.40 (td, J = 8.6, 6.9 Hz, 1H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1H), 7.11-7.01 (m, 1H), 5.39 (dd, J = 4.1, 10.4 Hz, 1H), 4.89 (dd, J = 4.2, 12.3 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1H), 4.36-^.22 (m, 1H)1 4.00 (dd, J = 10.2, 12.3 Hz, 1H), 3.67 (dd, J = 6.7, 8.6 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H).
Example Ae
After dissolution of 16.0 g of compound 17 (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml_ of 1N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml_ of EtOH and drying provided 13.5 g of compound 18 (80% yield) as a crystal.
1H NMR(300 MHz, DMSO-cfe) δ 10.73 (t, J = 6.0 Hz, 1H), 7.89 (s, 1H), 7.40-7.30 (m, 1H), 7.25-7.16 (m, 1H), 7.07-6.98 (m, 1H), 5.21 (dd, J = 3.8, 10.0 Hz, 1H), 4.58 (dd, J = 3.8, 12.1 Hz, 1H), 4.51 (d, J = 5.4 Hz, 2H), 4.3CM.20 (m, 2H), 3.75 (dd, J = 10.0, 12.1 Hz, 1H), 3.65-3.55 (m, 1H), 1.27 (d, J = 6.1 Hz, 3H).
………………
WO2010011814
http://www.google.st/patents/WO2010011814A1?cl=en&hl=pt-PT
Scheme 1
2a 2b
Scheme 2
Scheme 3
Scheme 4
phosphorylation
Scheme 5
Hydrogenolysis
The following examples are intended for illustratation only and are not intended to limit the scope of the invention in any way. Preparation 1 : (3S.11 af?VΛ/-r(2.4-DifluoroDhenvnmethyll-6-hvdroxy-3-methyl-5.7-dioxo- 2,3,5,7, 11 ,11 a-hexahydroM ,31oxazolor3,2-alpyridori ,2-c/1pyrazine-8-carboxamide sodium salt (compound 1 b, scheme 2).
I) MsCI, Et3N
2) DBU
P-1 P-2 P-3
a) Synthesis of 2-methyl-3-[(phenylmethvl)oxvl-4/-/-pvran-4-one (compound P-2). To a slurry of 2000 g of compound P-1(1.0 eq.) in 14.0 L of MeCN were added 2848 g of benzyl bromide(1.05 eq.) and 2630 g of K2CO3(1.2 eq.). The mixture was stirred at 80 0C for 5 h and cooled to 13°C. Precipitate was filtered and washed with 5.0 L of MeCN. The filtrate was concentrated and 3.0 L of THF was added to the residue. The THF solution was concentrated to give 3585 g of crude compound P-2 as oil. Without further purification, compound P-2 was used in the next step. 1H NMR(300 MHz, CDCI3) δ 7.60 (d, J = 5.7 Hz, 1 H), 7.4-7.3 (m, 5H), 6.37 (d, J = 5.7 Hz, 1 H), 5.17 (s, 2H), 2.09 (s, 3H).
b) Synthesis of 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound P-3). To 904 g of the crude compound P-2 was added 5.88 L of THF and the solution was cooled to -60 0C. 5.00 L of 1.0 M of Lithium bis(trimethylsilylamide) in THF(1.25 eq.) was added dropwise for 2 h to the solution of compound 2 at -60 0C. Then, a solution of 509 g of benzaldehyde(1.2 eq.) in 800 ml. of THF was added at -60 0C and the reaction mixture was aged at -60 0C for 1 h. The THF solution was poured into a mixture of 1.21 L of conc.HCI, 8.14 L of ice water and 4.52 L of EtOAc at less than 2 0C.
The organic layer was washed with 2.71 L of brine (twice) and the aqueous layer was extracted with 3.98 L of EtOAc. The combined organic layers were concentrated. To the mixture, 1.63 L of toluene was added and concentrated (twice) to provide toluene slurry of compound P-3. Filtration, washing with 0.90 L of cold toluene and drying afforded 955 g of compound P-3 (74% yield from compound P-1 ) as a solid. 1H NMR(300 MHz, CDCI3) δ
7.62 (d, J = 5.7 Hz, 1 H), 7.5-7.2 (m, 10H), 6.38 (d, J = 5.7 Hz, 1 H), 5.16 (d, J = 11.4 Hz, 1 H), 5.09 (d, J = 11.4 Hz, 1 H), 4.95 (dd, J = 4.8, 9.0 Hz, 1 H), 3.01 (dd, J = 9.0, 14.1 Hz, 1 H), 2.84 (dd, J = 4.8, 14.1 Hz, 1 H).
c) Synthesis of 2-[(£)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound
P-4). To a solution of 882 g of compound P-3 (1.0 eq.) in 8.82 L of THF were added 416 g of Et3N(1.5 eq.) and 408 g of methanesulfonyl chloride(1.3 eq.) at less than 30 0C. After confirmation of disappearance of compound P-3, 440 ml. of NMP and 1167 g of DBU(2.8 eq.) were added to the reaction mixture at less than 30 0C and the reaction mixture was aged for 30 min. The mixture was neutralized with 1.76 L of 16% sulfuric acid and the organic layer was washed with 1.76 L of 2% Na2S03aq. After concentration of the organic layer, 4.41 L of toluene was added and the mixture was concentrated (tree times). After addition of 4.67 L of hexane, the mixture was cooled with ice bath. Filtration, washing with 1.77 L of hexane and drying provided 780 g of compound P-4 (94% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.69 (d, J = 5.7 Hz, 1 H), 7.50-7.25 (m, 10H), 7.22 (d, J = 16.2
Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.41 (d, J = 5.7 Hz, 1 H), 5.27 (s, 2H). d) Synthesis of 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid (compound P-5). To a mixture of 822 g of compound P-4 (1.0 eq.) and 1 1.2 g of RuCI3-nH2O(0.02 eq.) in 2.47 L of MeCN, 2.47 L of EtOAc and 2.47 L of H2O was added 2310 g of NalO4(4.0 eq.) at less than 25 0C. After aging for 1 h, 733 g of NaCIO2(S-O eq.) was added to the mixture at less than 25 0C. After aging for 1 h, precipitate was filtered and washed with 8.22 L of
EtOAc. To the filtrate, 1.64 L of 50% Na2S203aq, 822 ml. of H2O and 630 ml. of coc.HCI were added. The aqueous layer was extracted with 4.11 L of EtOAc and the organic layers were combined and concentrated. To the residue, 4 L of toluene was added and the mixture was concentrated and cooled with ice bath. Filtration, washing with 1 L of toluene and drying provided 372 g of compound P-5 (56% yield) as a solid. 1H NMR(300 MHz,
CDCI3) δ 7.78 (d, J = 5.7 Hz, 1 H), 7.54-7.46 (m, 2H), 7.40-7.26 (m, 3H), 6.48 (d, J = 5.7 Hz, 1 H), 5.6 (brs, 1 H), 5.31 (s, 2H).
e) Synthesis of 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylic acid (compound P-6). A mixture of 509 g of compound P-5 (1.0 eq.) and
407 g of 3-amino-propane-1 ,2-diol(2.5 eq.) in 1.53 L of EtOH was stirred at 65 0C for 1 h and at 80 0C for 6 h. After addition of 18.8 g of 3-Amino-propane-1 ,2-diol(0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 1 h. After addition of 18.8 g of 3-amino- propane-1 ,2-diol (0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 30 min. After cooling and addition of 509 ml. of H2O, the mixture was concentrated. To the residue,
2.54 L of H2O and 2.54 L of AcOEt were added. After separation, the aqueous layer was washed with 1.02 L of EtOAc. To the aqueous layer, 2.03 L of 12% sulfuric acid was added at less than 12 0C to give crystal of compound P-6. Filtration, washing with 1.53 L of cold H2O and drying provided 576 g of compound P-6 (83% yield) as a solid. 1H NMR(300 MHz, DMSO-de) δ 7.67 (d, J = 7.5 Hz, 1 H), 7.5-7.2 (m, 5H), 6.40 (d, J = 7.5 Hz, 1 H), 5.07
(s, 2H), 4.2-4.0 (m, 1 H), 3.9-3.6 (m, 2H), 3.38 (dd, J = 4.2, 10.8 Hz, 1 H), 3.27 (dd, J = 6.0, 10.8 Hz, 1 H).
f) Synthesis of methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-7). To a slurry of 576 g of compound P-6 (1.0 eq.: 5.8% of H2O was contained) in 2.88 L of NMP were added 431 g of NaHCO3(3.0 eq.) and 160 ml. of methyl iodide(1.5 eq.) and the mixture was stirred at room temperature for 4 h. After cooling to 5 0C, 1.71 L of 2N HCI and 1.15 L of 20% NaClaq were added to the mixture at less than 10 0C to give crystal of compound 7. Filtration, washing with 1.73 L of H2O and drying provided 507 g of compound P-7 (89% yield) as a solid. 1H NMR(300 MHz, DMSO- cfe) δ 7.59 (d, J = 7.5 Hz, 1 H), 7.40-7.28 (m, 5H), 6.28 (d, J = 7.5 Hz, 1 H), 5.21 (d, J = 5.4 Hz, 1 H), 5.12 (d, J = 10.8 Hz, 1 H), 5.07 (d, J = 10.8 Hz, 1 H), 4.83 (t, J = 5.7 Hz, 1 H), 3.97 (dd, J = 2.4, 14.1 Hz, 1 H), 3.79 (s, 3H), 3.70 (dd, J = 9.0, 14.4 Hz, 1 H), 3.65-3.50 (m, 1 H), 3.40-3.28 (m, 1 H), 3.26-3.14 (m, 1 H).
g) Synthesis of methyl 1-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-8). To a mixture of 507 g of compound P -7 (1.0 eq.) in
5.07 L of MeCN, 5.07 L of H2O and 9.13 g of AcOH(0.1 eq.) was added 390 g of NaIO4(1.2 eq.) and the mixture was stirred at room temperature for 2 h. After addition of 1.52 L of 10% Na2S2OsBq., the mixture was concentrated and cooled to 10 0C. Filtration, washing with H2O and drying provided 386 g of compound P-8 (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.42-7.30 (m, 5H), 6.33 (d, J = 6.0 Hz, 2H),
6.29 (d, J = 7.5 Hz, 1 H), 5.08 (s, 2H), 4.95-4.85 (m, 1 H), 3.80 (s, 3H), 3.74 (d, J = 5.1 Hz, 2H).
h) Synthesis of (3S, 11 aR)-3-methyl-6-[(phenylmethyl)oxy]-2,3, 1 1 ,1 1a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-9). After dissolution of mixture of 320 g of compound P-8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 ml. of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 0C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 ml. of cold 2-propanol and drying provided 167 g of compound P-9 (52% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.23 (d, J = 10.2 Hz, 1 H), 5.20 (dd, J = 3.9, 9.6 Hz, 1 H), 4.46-4.34 (m, 1 H), 4.31 (dd, J = 6.6, 8.7 Hz, 1 H), 4.14 (dd, J = 3.9, 12.3 Hz, 1 H), 3.79 (dd, J = 9.9, 12.3 Hz, 1 H), 3.62 (dd, J = 6.9, 8.7 Hz, 1 H), 1.38 (d, J = 6.3 Hz, 3H).
i) Synthesis of (3 S, 1 1 aR)-8-bromo-3-methyl-6-[(phenylmethyl)oxy]-2,3, 11 ,11a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-10). To slurry of 156 g of compound P-9 (1.0 eq.) in 780 ml. of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound P-10 (84% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 8.37 (s, 1 H), 7.55- 7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1 H), 5.18 (d, J = 10.8 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.53 (dd, J = 3.6, 12.0 Hz, 1 H), 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 1 1.7 Hz, 1 H), 3.64 (dd, J = 5.7, 8.1 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3H). j) Synthesis of (3S,1 1aS)-Λ/-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,3,5,7, 11 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8- carboxamide (compound P-11). Under carbon mono-oxide atmosphere, a mixture of 163 g of compound P-10 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 mL of 2,4- difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 1 1.3 g of Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O, the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt.
The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound P-11 (96% yield) as foam. 1H NMR(300 MHz, CDCI3) δ 10.38 (t, J = 6.3 Hz, 1 H), 8.39 (s, 1 H), 7.75-7.25 (m, 7H), 6.90-6.70 (m, 2H), 5.43 (d, J = 10.2 Hz, 1 H), 5.24 (d, J = 10.2 Hz, 1 H), 5.19 (dd, J = 3.9, 9.9 Hz, 1 H), 4.63 (d, J = 6.0 Hz, 2H), 4.50-4.25 (m, 3H), 3.86 (dd, J = 9.9, 12.3 Hz, 1 H), 3.66 (dd, J = 6.9, 8.4 Hz,
1 H), 1.39 (d, J = 6.0 Hz, 3H).
k) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide (compound 1a). Under hydrogen atmosphere, a mixture of 184 g of compound P-11 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1 ), the filtrate was concentrated. After 200 mL of AcOEt was added to the residue, filtration afforded crude solid of compound 1 a.
The precipitates were combined and extracted with 4.0 L of CHCI3/Me0H(5/1 ). After concentration of the CHCI3/MeOH solution and addition of 250 mL of AcOEt to the residue, filtration afforded crude solid of compound 1a. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated
(three times). After cooling of the residue, filtration and drying provided 132 g of compound 1a (88% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 11.47 (brs, 1 H), 10.31 (t, J = 6.0 Hz, 1 H), 8.46 (s, 1 H), 7.40 (td, J = 8.6, 6.9 Hz, 1 H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1 H), 7.11- 7.01 (m, 1 H), 5.39 (dd, J = 4.1 , 10.4 Hz, 1 H), 4.89 (dd, J = 4.2, 12.3 Hz, 1 H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1 H), 4.36-4.22 (m, 1 H), 4.00 (dd, J = 10.2, 12.3 Hz, 1 H), 3.67 (dd, J = 6.7, 8.6 Hz, 1 H), 1.34 (d, J = 6.3 Hz, 3H).
I) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide sodium salt (compound 1 b). After dissolution of 16.0 g of compound 1a (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml. of 1 N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml. of EtOH and drying provided 13.5 g of compound 1b (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 10.73 (t, J = 6.0 Hz, 1 H), 7.89 (s, 1 H), 7.40-7.30 (m, 1 H), 7.25-7.16 (m, 1 H), 7.07-6.98 (m, 1 H), 5.21 (dd, J = 3.8, 10.0 Hz, 1 H), 4.58 (dd, J = 3.8, 12.1 Hz, 1 H), 4.51 (d, J = 5.4 Hz, 2H), 4.30-4.20 (m, 2H), 3.75 (dd, J = 10.0, 12.1 Hz, 1 H), 3.65-3.55 (m, 1 H), 1.27 (d, J = 6.1 Hz, 3H).
References
- PrEP GSK744 Integrase Administered Monthly Perhaps Quarterly Prevents HIV-Infection in Monkeys. 20th Conference on Retroviruses and Opportunistic Infections. Atlanta, GA March 3–6, 2013.
- http://www.natap.org/2013/CROI/croi_38.htm
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2006116764A1 * |
Apr 28, 2006 |
Nov 2, 2006 |
Shionogi & Co |
Polycyclic carbamoylpyridone derivative having hiv integrase inhibitory activity |
| US6919351 * |
Oct 9, 2001 |
Jul 19, 2005 |
Merck & Co., Inc. |
Aza-and polyaza-naphthalenyl-carboxamides useful as HIV integrase inhibitors |
| WO2012018065A1 * |
Aug 4, 2011 |
Feb 9, 2012 |
Shionogi & Co., Ltd. |
Process for preparing compound having hiv integrase inhibitory activity |
| WO2012151361A1 |
May 3, 2012 |
Nov 8, 2012 |
Concert Pharmaceuticals Inc. |
Carbamoylpyridone derivatives |
| WO2013038407A1 * |
Sep 2, 2012 |
Mar 21, 2013 |
Mapi Pharma Ltd. |
Amorphous form of dolutegravir |
| US8552187 |
Dec 9, 2009 |
Oct 8, 2013 |
Shionogi & Co., Ltd. |
Processes and intermediates for carbamoylpyridone HIV integrase inhibitors |
| US8580967 |
Jul 23, 2009 |
Nov 12, 2013 |
Shionogi & Co., Ltd. |
Methyl 3-(benzyloxy)-1-(2,2-dihydroxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylate and processes for the preparation thereof |
| US8624023 |
Dec 8, 2009 |
Jan 7, 2014 |
Shionogi & Co., Ltd. |
Synthesis of carbamoylpyridone HIV integrase inhibitors and intermediates |
Filed under:
Phase2 drugs,
Uncategorized Tagged:
CARBOTEGRAVIR,
GSK 744,
hiv,
phase 2 ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()



 The experimental drug was developed by Prof Frank Longo from Stanford University
The experimental drug was developed by Prof Frank Longo from Stanford University






















 56 and the triplet at
56 and the triplet at 














 14 and the triplet at
14 and the triplet at 
















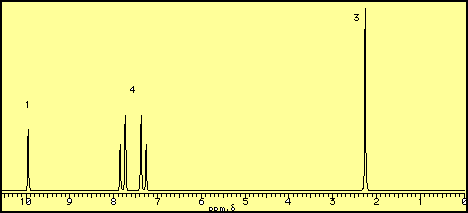









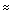 1 Hz) with the peak at
1 Hz) with the peak at 

 methyl vinyl ether (methyl ethenyl ether)
methyl vinyl ether (methyl ethenyl ether)

 Name: 1,2-dimethoxyethane
Name: 1,2-dimethoxyethane

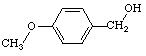
 4-isopropyl-1-methoxybenzene (4-(1-methylethyl)-1-methoxybenzene)
4-isopropyl-1-methoxybenzene (4-(1-methylethyl)-1-methoxybenzene)

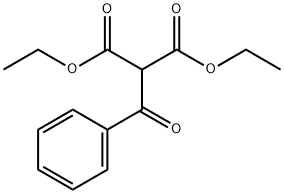
 CH(CH3) 2 in which the carbon is bonded to something mildly electronegative and the two doublets centered at
CH(CH3) 2 in which the carbon is bonded to something mildly electronegative and the two doublets centered at 
 3-bromomethyltoluene (3-bromomethyl-1-methylbenzene)
3-bromomethyltoluene (3-bromomethyl-1-methylbenzene)














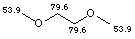
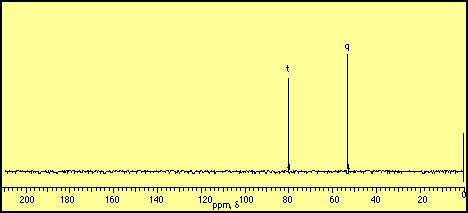




 CH- group. The remaining CH2 group at
CH- group. The remaining CH2 group at 



 -system of alkenes, aromatic compounds and carbonyls strongly deshield
-system of alkenes, aromatic compounds and carbonyls strongly deshield 
 C nuclei and move them “downfield” to higher ppm values.
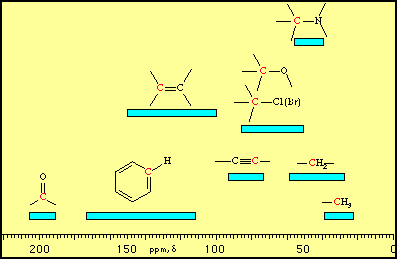
C nuclei and move them “downfield” to higher ppm values.  Simple interior (primary and secondary) carbons tend to be in the range
Simple interior (primary and secondary) carbons tend to be in the range  ) position due to gauche interactions. This is shown schematically below and the gamma position is marked above in the example for hexane.
) position due to gauche interactions. This is shown schematically below and the gamma position is marked above in the example for hexane. The presence of an electronegative atom such as oxygen tends to move the chemical shift of the Œ-carbon down into the region
The presence of an electronegative atom such as oxygen tends to move the chemical shift of the Œ-carbon down into the region  Halogens, however, have effects which are difficult to predict and carbons adjacent to halogens tend to have chemical shifts in the
Halogens, however, have effects which are difficult to predict and carbons adjacent to halogens tend to have chemical shifts in the  Alkene carbons tend to have chemical shifts in the range
Alkene carbons tend to have chemical shifts in the range 
 Alkyne carbons occur in the region
Alkyne carbons occur in the region  Carbonyls are the most highly deshielded carbons which are typically encountered. Their intensity is usually weak, since there are no attached hydrogens to contribute to the Nuclear Overhauser Effect enhancement (with the exception of aldehydes). Typical chemical shifts occur in the region
Carbonyls are the most highly deshielded carbons which are typically encountered. Their intensity is usually weak, since there are no attached hydrogens to contribute to the Nuclear Overhauser Effect enhancement (with the exception of aldehydes). Typical chemical shifts occur in the region  Aromatic carbons have chemical shifts in the range
Aromatic carbons have chemical shifts in the range 






































 Originally posted on
Originally posted on 
























![VTO=\frac{\text{nominal volume of all reactors} [m^3]*\text{time per batch} [h]}{\text{output per step} [kg]}](http://upload.wikimedia.org/math/7/9/a/79aca4fd95405c01c04df9181fb3ee63.png)
















 Photo by Rob Hill/MU Publications
Photo by Rob Hill/MU Publications

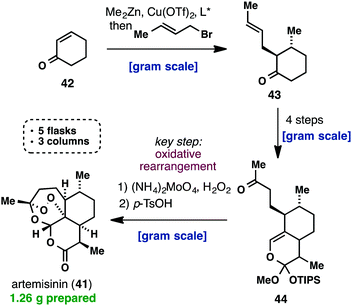













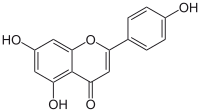 apigenin
apigenin







 Originally posted on
Originally posted on 















 http://www.ncbi.nlm.nih.gov/pubmed/24568529″ width=”395″ height=”395″ />
http://www.ncbi.nlm.nih.gov/pubmed/24568529″ width=”395″ height=”395″ />











































































































































































































 Botanical Source:Cuscuta chinensis(The ripe seed of Cuscuta chinensis Lam.,an annual voluble parasitic herb of the family Convolvulaceae).
Botanical Source:Cuscuta chinensis(The ripe seed of Cuscuta chinensis Lam.,an annual voluble parasitic herb of the family Convolvulaceae).








