![File:Prulifloxacin.png]()
PRULIFLOXACIN
(RS)-6-Fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl]-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid
6-Fluoro-1-methyl-7-(4-(5-methyl-2-oxo-1,3-dioxelen-4-yl)methyl-1-piperazinyl)-4-oxo-4H-(1,3)thiazeto(3,2-a)quinoline-3-carboxylic acid
123447-62-1 CAS NO
NM 441, Quisnon, Pruvel, Sword, Prulifloxacin [INN], 123447-62-1, NM-441, CCRIS 7686, NCGC00164615-01NAD-441A
OPT-99
Molecular Formula: C21H20FN3O6S
Molecular Weight: 461.463403
Launched – 2002 BY NIPPON SHINYAKU
SYNTHESIS…….http://www.drugfuture.com/synth/syndata.aspx?ID=151640
Prulifloxacin is an older synthetic chemotherapeutic antibiotic of the fluoroquinolone drug class[1][2] undergoing clinical trials prior to a possible NDA (New Drug Application) submission to the U.S. Food and Drug Administration (FDA). It is a prodrug which is metabolized in the body to the active compound ulifloxacin.[3][4] It was developed over two decades ago by Nippon Shinyaku Co. and was patented in Japan in 1987 and in the United States in 1989.[5][6]
It has been approved for the treatment of uncomplicated and complicated urinary tract infections, community-acquired respiratory tract infections in Italy and gastroenteritis, including infectious diarrheas, in Japan.[3][7] Prulifloxacin has not been approved for use in the United States.
Prulifloxacin is a novel fluoroquinolone antibiotic that was launched pursuant to a collaboration between Meiji Seika and Nippon Shinyaku in 2002 for the oral treatment of systemic bacterial infections, including acute upper respiratory tract infection, bacterial pneumonia, prostatitis, cholecystitis, bacterial enteritis, internal genital infections, otitis media, sinusitis and others. It is currently marketed in a tablet formulation. A once-daily formulation to be taken over a three-day period is in phase III clinical trials at Optimer Pharmaceuticals to be used in the treatment of bacterial gastroenteritis, including traveler’s diarrhea. The formulation had been in phase II trials at the company for the treatment of urinary tract infections, however, no recent development for this indication have been reported. The drug has also been studied at Optimer for the treatment of community-acquired respiratory tract infections, but recent progress reports for this indication have not been made available.
Prulifloxacin has in vitro activity against a wide range of gram-negative and gram-positive microorganisms. Its antibacterial action results from inhibition of DNA gyrase and topoisomerase IV, both Type II isomerases. DNA gyrase is an essential enzyme that is involved in the replication, transcription, and repair of bacterial DNA. Topoisomerase IV is an enzyme known to play a key role in the partitioning of the chromosomal DNA during bacterial cell division. Together, the Type II topoisomerases remove the positive supercoils that accumulate ahead of a translocating DNA polymerase, allowing DNA replication to continue unhindered by topological strain. Fluoroquinolones may be active against pathogens that are resistant to penicillins, cephalosporins, aminoglycosides, macrolides and tetracyclines, as they possess a distinct mechanism of action from these antibiotics.
Prulifloxacin was discovered by Nippon Shinyaku and codeveloped with Meiji Seika in Japan. Nippon Shinyaku granted Angelini a manufacturing and marketing license for Italy in 1993. Exclusive Korean manufacturing and commercialization rights were acquired by Yuhan from Nippon Shinyaku in March 2003. In June 2004, Optimer was granted exclusive development and commercialization rights to prulifloxacin in the U.S. from Nippon Shinyaku. Finally, Recordati signed a nonexclusive licensing agreement with Angelini for the marketing and sale of prulifloxacin in Spain in October 2004. In March 2009, the product was licensed to Lee’s Pharmaceuticals by Nippon Shinyaku for marketing in China as an oral treatment of bacterial infection. In 2010, prulifloxacin was licensed to Algorithm by Nippon Shinyaku in North Africa and the Middle East for the development and marketing for the treatment of bacterial infections.
History
In 1987 a European Patent (EP 315828) for prulifloxacin (Quisnon ) was issued to the Japanese based pharmaceutical company, Nippon Shinyaku Co., Ltd (Nippon). Ten years after the issuance of the European patent, marketing approval was applied for and granted in Japan (March 1997). Subsequent to being approved by the Japanese authorities in 1997 prulifloxacin (Quisnon) was co-marketed and jointly developed in Japan with Meiji Seika as licensee (Sword).[6]
In more recent times, Angelini ACRAF SpA, under license from Nippon Shinyaku, has fully developed prulifloxacin, for the European market.[8] Angelini is the licensee for the product in Italy. Following its launch in Italy, Angelini launched prulifloxacin in Portugal (January 2007) and it has been stated that further approvals will be sought in other European countries.[9][10]
Prulifloxacin is marketed in Japan and Italy as Quisnon (Nippon Shinyaku); Sword (Meiji); Unidrox (Angelini) and generic as Pruquin.
In 1989 and 1992 United States patents (US 5086049) were issued to Nippon Shinyaku for prulifloxacin. It was not until June 2004, when Optimer Pharmaceuticals acquired exclusive rights to discover, develop and commercialize prulifloxacin (Pruvel) in the U.S. from Nippon Shinyaku Co., Ltd., that there were any attempts to seek FDA approval to market the drug in the United States. Optimer Pharmaceuticals expects to file an NDA (new drug application) for prulifloxacin some time in 2010. As the patent for prulifloxacin has already expired, Optimer Pharmaceuticals has stated that this may have an effect on the commercial prospects of prulifloxacin within the United States market.[11]
Licensed uses
Prulifloxacin has been approved in Italy ,Japan,China,India and Greece (as indicated), for treatment of infections caused by susceptible bacteria, in the following conditions:
- Italy
- Acute uncomplicated lower urinary tract infections (simple cystitis)
- Complicated lower urinary tract infections
- Acute exacerbation of chronic bronchitis
- Japan
- Gastroenteritis, including infectious diarrheas
- Other countries
- Prulifloxacin has not been approved for use in the United States, but may have been approved in other Countries, other than that which is indicated above.
Availability
Prulifloxacin is available as:
- Tablets (250 mg, 450 mg or 600 mg)
In most countries, all formulations require a prescription.
![]()
Prulifloxacin is chemically known as 6-fluoro-1-methyl-7-{4-[(5-methyl-2-oxo-1 ,3-dioxol- 4-yl)methyl]piperazin-1-yl}-4-oxo-4H-[1 ,3]-thiazeto-[3,2-a]-quinoline-3-carboxylic acid, and it has the structure as shown below as formula I:
FORMULA I
Prulifloxacin has significant antibacterial activity and has been marketed as a synthetic antibacterial agent.
Prulifloxacin was first disclosed in US 5,086,049. The patent discloses a process for the preparation of prulifloxacin by the condensation of ulifloxacin with a 4-halomethyl-5- methyl-1 ,3-dioxolen-2-one of formula III
wherein X is halo selected form chloro, bromo or iodo, in the presence or absence of an aprotic solvent and a base to obtain prulifloxacin free base which is recrytallised with chloroform-methanol. In an exemplified process, ethyl 6,7-difluoro-1-methyl-4-oxo-4H- (1 ,3)-thiazeto-(3,2-a)-quinoline-3-carboxylate is condensed with piperazine in the presence of dimethyl formamide and purified by column chromatography followed by basic hydrolysis to give ulifloxacin, which is then converted to prulifloxacin.
The above process involves column chromatography. Prulifloxacin prepared by this method has a purity of 60-65% containing impurities in unacceptable levels. Removal of these impurities by usual purification procedures, such as recrystallisation, distillation and washing, is difficult and requires extensive and expensive multiple purification processes. This further decreases the overall yield. A method involving column chromatographic purifications and multiple purifications cannot be used for large-scale operations, thereby making the process commercially non-viable.
European Patent No. 315828 disclosed a variety of quinoline carboxylic acid derivatives and pharmaceutically acceptable salts thereof. These compounds are exhibiting antibacterial activity and useful as remedies for various infectious diseases. Among them prulifloxacin, chemically (+)-6-Fluoro- 1 -methyl-7-[4-(5-methyl-2-oxo-1 ,3-dioxolen-4-ylmethyl)-1 -piperazinyl]-4-oxo-4H- [1 ,3]thiazeto[3,2-a]quinoline-3-carboxylic acid is a fluoroquinolone antibacterial prodrug which shows potent and broad-spectrum antibacterial activity both in vitro and in vivo. Prulifloxacin also showed superior activity against strains of Enterobacteriaceae and Pseudomonas aeruginosa. Prulifloxacin is represented by the following structure:
Processes for the preparation of prulifloxacin and related compounds were disclosed in European Patent No. 315828 and UK Patent Application No. GB 2190376.
In – the preparation of prulifloxacin, 6-fluoro-1-methyl-4-oxo-7-(1- piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylic acid of formula I:
![Figure imgf000003_0001]()
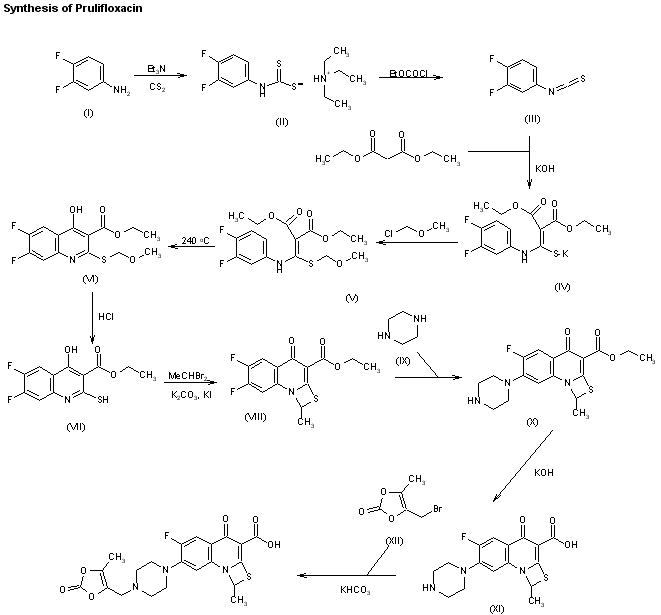
is a key intermediate. According to the UK Patent Application No. GB 2190376, the compound of the formula I was prepared by the reaction of 3,4-difluroaniline with carbon disulfide and triethylamine to give triethylammonium N-(3,4- difluorophenyl)dithio carbamate, which by reaction with ethyl chloroformate and triethylamine in chloroform is converted into 3,4-difluorophenyl isothiocyanate, followed by reaction with diethyl malonate and KOH in dioxane affords the potassium salt, which is then treated with methoxymethyl chloride in dimethylformamide to give diethyl 1-(3,4-difluorophenylamino)-1- (methoxymethylthio)-rnethylene-rnalonate. The cyclization of the thio compound at 2400C in diphenyl ether affords ethyl 6,7-difluoro-4-hydroxy-2- methoxymethylthioquinoline-3-carboxylate, which by treatment with HCI in ethanol gives ethyl δy-difluoro^-hydroxy^-mercaptoquinoline-S-carboxylate. The cyclization of the mercapto compound with 1,1-dibromoethane by means of potassium carbonate and potassium iodide in hot dimethylformamide yields ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate, which is condensed with piperazine in dimethylformamide to afford ethyl 6- fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3- carboxylate, which is then subjected to hydrolysis with potassium hydroxide in hot tert-butanol to give the compound of formula I.
The compound of formula I obtained by the process described in the UK Patent Application No. GB 2190376 is not satisfactory from purity point of view, the reaction between ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2- a]quinoline-3-carboxylate and piperazine requires longer time about 48 hours to complete, the yield obtained is not satisfactory, and the process also involves column chromatographic purifications. Methods involving column chromatographic purifications cannot be used for large-scale operations, thereby making the process commercially not viable. According to the European Patent No. 315828, prulifloxacin is prepared by reacting 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a] quinoline-3-carboxylic acid with 4-bromomethyl-5-methyl-1 ,3-dioxolen-2-one in presence of potassium bicarbonate in dimethylformamide. However, a need still remains for an improved and commercially viable process of preparing pure prulifloxacin that will solve the aforesaid problems associated with process described in the prior art and will be suitable for large- scale preparation, in terms of simplicity, purity and yield of the product.
Prulifloxacin is chemically 6-fluoro-l-methyl-7-{4-[(5-methyl-2-oxo-l,3-dioxol-4- yl)methyl]piperazin-l-yl}-4-oxo-4H-[l,3]thiazeto[3,2-α]quinoline-3-carboxylic acid of Formula I having the structure as depicted below:
FORMULA I
Prulifloxacin has significant antibacterial activity and has been marketed as a synthetic antibacterial agent. U.S. Patent No. 5,086,049 provides a process for the preparation of prulifloxacin by reacting 6-fluoro-l-methyl-4-oxo-7-piperazin-l-yl-4H- [l,3]thiazeto[3,2-α]quinoline-3-carboxylic acid of Formula II,
FORMULA II and 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one of Formula III,
FORMULA III using N,N-dimethylformamide as a solvent. 4-(Bromomethyl)-5-methyl-l,3-dioxol-2-one of Formula III is used in excess to one mole of the compound of Formula II. The process provided in U.S. Patent No. 5,086,049 further involves concentrating the reaction mixture, pouring the residue into water and isolating prulifloxacin by filtration. The resulting prulifloxacin is recrystallized from chloroform-methanol.
However, U.S. Patent No. 5,086,049 does not provide any method to remove the unreacted or the excess of 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one of Formula III used as a starting material. The present inventors have observed that it is difficult to obtain prulifloxacin with pharmaceutically acceptable purity by following the process provided in U.S. Patent No. 5,086,049, which is typically contaminated by process related impurities including 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one
A need still remains for an improved and commercially-viable process for preparing pure prulifloxacin that will solve the aforesaid problems associated with the process described in the prior art and that will be suitable for large-scale preparation, in terms of simplicity, purity and yield of the product.
EP1626051 A1 mentions that Type I, Type II and Type III crystals of prulifloxacin are obtained by crystallization from acetonitrile as reported in lyakuhin Kenkyu, Vol. 28 (1), (1997), 1-11. However, the conditions of crystallization from acetonitrile for preparing Type I, Type II and Type III crystals are not disclosed in lyakuhin Kenkyu, Vol. 28 (1), (1997), 1-11. EP1626051A1 further mentions that Type III crystals have been marketed by considering the solubility, absorbability, therapeutic effect and the like of the respective crystal forms.
US 2007/0149540 discloses a crystal of prulifloxacin acetonitrile solvate (Compound B) which is an intermediate for producing preferentially the type III crystal of prulifloxacin. A crystal of Compound B can be preferentially precipitated by controlling the supersaturation concentration in crystallization using acetonitrile as a solvent, subsequently; the type III crystal of Compound A can be produced by performing desolvation of the crystal.
WO 2008/111018 discloses processes for the preparation of Type I, Type II and Type III crystals of prulifloxacin. There is disclosed a process for preparing Type I crystals by controlled cooling over a period of 7 to 9 hours and prolonged drying over 24 hours. The inventors of the present invention have found that Type I and Type III crystals prepared according to the WO 2008/111018 process are unstable and the process is non-reproducible.
WO 2010/0084508 discloses processes for the preparation of Type I, Type II and Type III crystals of prulifloxacin.
WO 2008/059512 discloses a process for the preparation of prulifloxacin using novel intermediates.
WO 2008/111016 discloses a process for the preparation of prulifloxacin having purity of about 99% or above. It would be a significant contribution to the art to provide a crystalline form of prulifloxacin, which is consistent and to provide industrially viable methods of preparation, pharmaceutical formulations, and methods of use thereof.
![]()
…………………
SYNTHESIS
http://www.google.com/patents/WO2012001357A1?cl=en
Scheme 1.
Formula I
[PRULIFLOXACIN]
Example 1
Preparation of ethyl-6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-(1 ,3)-thiazeto-[3,2-a]- quinoline-3-carboxylate (formula III)
5,6-difluoro-1-methyl-4-oxo-4H-[1 ,3]-thiazeto-[3,2-a]-quinoline-3-carboxylic acid ethyl ester of formula (II) (100 gms, 0.321 moles) was stirred in 500 ml of DMF at room temperature. Piperazine (76 gms, 0.882 moles) was added at room temperature and stirred for 10 minutes. The temperature was slowly raised to 50-55°C and the reaction mass was stirred at 50-55°C for 5 hours. After completion of the reaction, the reaction mass was cooled to 25-30°C and stirred for 2 hours. The reaction mass was further chilled to 10-15°C and stirred for 2 hours. The precipitated solid was filtered, washed of chilled DMF (2 x 50 ml). The solid was slurry washed with water (300 ml), filtered, washed with water ( 2 x 100 ml) and dried under vacuum at 70-75°C to yield the title compound [90 gms, 74 % yield, 95% HPLC purity].
Example 2
Preparation of 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]-thiazeto-[3,2-a]- quinoline-3-carboxylic acid (formula IV)
Ethyl-6-fluoro-1 -methyl-4-oxo-7-(1 -piperazinyl)-4H-(1 ,3)-thiazeto-[3,2-a]-quinoline-3- carboxylate (100 gms, 0.265 moles) was stirred in water (600 ml) at 25-30°C. To this potassium hydroxide solution (50 gms of potassium hydroxide flakes is dissolved in 200 ml of water) was added and the reaction mass was heated to 80-85°C. The contents were stirred for 1 hour and after completion of reaction, the reaction mass was cooled to 25-30°C. The pH of the reaction mass was adjusted to 6.5-7.0 using 1:1 aqueous acetic acid solution. The contents were stirred at room temperature for 1 hour. The precipitated solid was filtered, washed with water (2 x 100 ml). The solid was slurried in methanol (300 ml) for 1 hour at 25-30°C, filtered, washed with methanol (2 x 50 ml) and dried under vacuum at 70-75°C to yield the title compound [90 gms, 97% yield, 96% HPLC purity]. Example 3
Preparation of prulifloxacin
To a solution of 4-(chloromethyl)-5-methyl-1,3-dioxol-2-one (55 gms, 0.371 moles) in 50 ml of DMF at 25-30°C, sodium bromide (77 gms, 0.748 moles) was added and the reaction mass was slowly heated to 40-45°C. The contents were stirred at 40-45°C for 2 hours, acetone ( 500 ml) was added at 40-45°C and stirred for 3 hours. The reaction mass was filtered over hyflo, and the bed washed with acetone (100 ml). The solvent was completely distilled off under vacuum below 45°C to yield 4-(bromomethyl)-5- methyl-1 ,3-dioxol-2-one (formula V).
To a solution of 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]-thiazeto-[3,2-a]- quinoline-3-carboxylic acid of formula IV (100 gms, 0.286 moles) in 4.0 It of acetonitrile, DIPEA (70 ml , 0.402 moles)) was added at room temperature, stirred for 10 minutes. The reaction mass was cooled to 10-15°C and a solution of 4-(bromomethyl)-5-methyl- 1 ,3-dioxol-2-one (formula V) in 500 ml of acetonitrile was slowly added at 10-15°C over a period of 1 hour. The contents were stirred at 25-30°C for 20 hour, filtered over hyflo, and the bed washed with 200 ml of acetonitrile. The solvent was distilled off completely under vacuum below 50°C. Acetonitrile (100 ml) was added at 50°C and the contents were stirred for 30-60 minutes. The reaction mass was slowly chilled to 0-5°C and the precipitated solid was filtered, washed with acetonitrile (25 ml) and dried to yield 65 gms of prulifloxacin. Example 4
Preparation of Type I crystals of prulifloxacin
Prulifloxacin (65 gms) was added to 200 ml of DMF at 25-30°C and heated to 80-85°C for 1 hour. The mixture was then slowly cooled to 25-30°C, stirred for 2 hours, chilled to 0-5°C for 2 hours. The precipitated solid was filtered and dried under vacuum at 70- 75°C to yield Type I crystals of prulifloxacin (55 gms, 99.5 % HPLC purity).
Example 5
Preparation of prulifloxacin
(55 gms, 0.371 moles) of 4-(chloromethyl)-5-methyl-1 ,3-dioxol-2-one is taken in 5.0 ml of DMF at 25-30°C. (77 gms, 0.748 moles) of sodium bromide is added and slowly heated the reaction mass to 40-45°C. The contents are stirred at 40-45°C for 2 hours, 500 ml of acetone is added at 40-45°C and stirred for 3 hours. The reaction mass is clarified over hyflo, and the bed washed with 100 ml of acetone to yield a solution of 4- (bromomethyl)-5-methyl-1 ,3-dioxol-2-one (formula V).
To a solution of 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]-thiazeto-[3,2-a]- quinoline-3-carboxylic acid of formula IV (100 gms, 0.286 moles) in 3.5 Its of acetone was at room temperature DIPEA (70 ml, 0.402 moles) and stirred for 10 minutes. The reaction mass was cooled to 10-15°C and a solution of 4-(bromomethyl)-5-methyl-1 ,3- dioxol-2-one (formula V) in acetone was slowly added to the reaction mass at 10-15°C over a period of 1 hour. The contents were further stirred at 25-30°C for 20 hour, filtered over hyflo and the bed washed with 200 ml of acetone. The solvent was distilled off completely under vacuum below 50°C. Acetonitrile (100 ml) was added at 50°C and the contents were stirred for 30-60 minutes. The reaction mass was further chilled to 0- 5°C and stirred for 2 hours. The precipitated solid was filteredand dried to yield prulifloxacin.
………………….
http://www.google.com/patents/WO2008059512A1?cl=en
novel process for preparing 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto [3,2-a]quinoline-3-carboxylic acid of formula I:
which comprises: a) reacting the difluoro-quinoline compound of formula
wherein R represents hydrogen atom or alkyl containing 1 to 4 carbon atoms; with boric acid of formula III:
in presence of acetic anhydride and acetic acid to give borane compound of formula IV:
b) reacting the borane compound of formula IV with piperazine of formula V:
HN NH V
to give piperazine compound of formula Vl:
c) treating the compound of formula Vl with an alkaline metal hydroxide, carbonate or bicarbonate to obtain the compound of formula I.
Prulifloxacin and pharmaceutically acceptable acid addition salts of prulifloxacin can be prepared by using the compound of formula I by known methods for example as described in the European Patent No. 315828. Borane compound of the formula IV and Vl are novel and forms part of the invention. Preferably the reaction in step (a) is carried out at about 300C to reflux temperature more preferably at about 800C to reflux temperature and still more preferably at reflux temperature.
Example 1 Step-I:
Acetic anhydride (24 ml) and acetic acid (11 ml) are added to boric acid (3.5 gm) under stirring at 25 – 300C, the contents are heated to reflux and then stirred for 3 hours at reflux. The reaction mass is cooled to 1000C, ethyl 6,7- difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate (20 gm) is added at 1000C, the contents are heated to reflux and then refluxed for 2 hours. The reaction mass is cooled to 25 – 350C, toluene (200 ml) is added under stirring, the reaction mass is cooled to 50C and then stirred for 1 hour at 5 – 100C. Filtered the solid, washed with 20 ml of toluene and then dried to give 25.5 gm of 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate -O3,04/bis/acetato-0/-borone. Step-I I: Acetonitrile (125 ml), dimethylsulfoxide (125 ml) and piperazine (13.8 gm) are added to 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3- carboxylate-03,04/bis/acetato-0/-borone (25.5 gm, obtained in step-l) under stirring at 25 – 350C, the contents are heated to 850C and then stirred for 3 hours at 80 – 850C to form a clear solution. The solution is cooled to 100C and then stirred for 1 hour at 10 – 150C. Filtered the solid, washed with 25 ml of acetonitrile and then dried to give 26 gm of 6-fluoro-1-methyl-4-oxo-7-(1- piperazinyl)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate-03,04/bis/acetato-0/- borone. Step-Ill: Water (155 ml), potassium hydroxide (17 gm) are added to 6-fluoro-1- methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate- O3,O4/bis/ acetato-0/-borone (26 gm, obtained in step-ll) under stirring at 25 – 350C, the contents are heated to 650C and then stirred for 4 hours at 60 – 650C. The reaction mass is cooled to 250C, filtered the undesired solid on hi-flow bed and then pH of the resulting filtrate is adjusted to 7 – 7.5 with 50% HCI solution at 25 – 300C. The separated solid is stirred for 1 hour at 25 – 300C, filtered the solid, washed with 35 ml of water and then dried to give 17 gm of 6-fluoro-1- methyl-4-oxo-7-(1 -piperazinyl)-4H-[1 ,3]thiazeto [3,2-a]quinoline-3-carboxylic acid (HPLC Purity: 98.5%). Example 2 Step-I:
Acetic anhydride (12 ml) and acetic acid (5.5 ml) are added to boric acid (1.25 gm) under stirring at 25 – 300C, the contents are heated to reflux and then stirred for 3 hours at reflux. The reaction mass is cooled to 1000C, 6,7-difluoro-1- methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylic acid (10 gm) is added at 1000C, the contents are heated to reflux and then refluxed for 3 hours. The reaction mass is cooled to 500C, toluene (100 ml) is added under stirring at 500C, the resulting mass is cooled to 100C and then stirred for 1 hour at 10 – 150C. Filtered the solid, washed with 20 ml of toluene and then dried to give 10 gm of 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate -O3 , 04/bis/acetato-0/-borone . Step-I I:
Acetonitrile (50 ml), dimethylsulfoxide (50 ml) and piperazine (5.5 gm) are added to 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3- carboxylate-03,04/bis/acetato-0/-borone (10 gm, obtained in step-l) under stirring at 25 – 350C, the contents are heated to 850C and then stirred for 3 hours at 80 – 850C to form a clear solution. The solution is cooled to 100C and then stirred for 1 hour at 10 – 150C. Filtered the solid, washed with 10 ml of acetonitrile and then dried to give 10.4 gm of 6-fluoro-1-methyl-4-oxo-7-(1- piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate-03,04/bis/acetato-0/- borone. Step-Ill :
Water (62 ml), potassium hydroxide (7 gm) are added to 6-fluoro-1-methyl-4- oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate-03,04/bis/ acetato-OAborone (10.4 gm, obtained in step-ll) under stirring at 25 – 350C, the contents are heated to 650C and then stirred for 4 hours at 60 – 650C. The reaction mass is cooled to 250C, filtered the undesired solid on hi-flow bed and then pH of the resulting filtrate is adjusted to 7 – 7.5 with 50% HCI solution at 25 – 300C. The separated solid is stirred for 30 minutes at 25 – 300C, filtered the solid, washed with 20 ml of water and then dried to give 68 gm of 6-fluoro-1- methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto [3,2-a]quinoline-3-carboxylic acid (HPLC Purity: 98.6%). Example 3
Acetonitrile (560 ml) and potassium bicarbonate (8 gm) are added to 6- fluoro-i-methyM-oxo-y-CI-piperazinyO^H-CI .SKhiazetofS^-alquinoline-S- carboxylic acid (14 gm, obtained as per the processes described in examples 1 and 2) under stirring at 25 – 300C, the contents are cooled to 150C and then the solution of 4-bromomethyl-5-methyl-1 ,3-dioxolen-2-one (10 gm) in acetonitrile (140 ml) is added at 15 – 200C for 30 to 45 minutes. The contents are stirred for 25 hours at 25 to 300C, filtered and the resulting filtrate is distilled under vacuum. To the residue added acetonitrile (70 ml), cooled the mass to 200C and then stirred for 1 hour to 1 hour 30 minutes at 20 – 250C. Filtered the solid, washed the solid with 15 ml of chilled acetonitrile and then dried to give 16 gm of prulifloxacin crude (HPLC Purity: 98.8%).
To the prulifloxacin crude (obtained above) added acetonitrile (200 ml) at 25 – 300C, the contents are heated to reflux and then refluxed for 30 minutes. To the reaction mass added activated carbon (5 gm) and refluxed for 15 minutes. The reaction mass is filtered on hi-flo bed, the resulting filtrate is cooled to 200C and then stirred for 3 – 4 hours at 20 – 250C. Filtered the solid, washed with 20 ml of acetonitrile and then dried to give 14 gm of prulifloxacin (HPLC Purity: 99.9%).
…………………
http://www.google.com/patents/WO2008111016A1?cl=en
In a first aspect, a process for the preparation of prulifloxacin is provided, the process comprising: a) reacting a compound of Formula II with a compound of Formula III to obtain prulifloxacin;
FORMULA III
FORMULA II
b) contacting the prulifloxacin obtained in step a) with an acid in a biphasic solvent system, wherein the biphasic solvent system comprises water and a water- immiscible organic solvent; c) separating the aqueous layer from the reaction mixture obtained in step b); d) treating the aqueous layer with a base; and e) isolating prulifloxacin.
The process described in steps b – e above may be carried out with prulifloxacin made from any process however.
The compounds of Formula II and Formula III may be prepared according to the methods provided in U.S. Patent No. 5,086,049.
Example 1: Process for the Preparation of Prulifloxacin:
Step A): A solution of 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one (35.5 g, 0.184 mole) in N,N-dimethylformamide (200 ml) was added dropwise at 0 to 5° C to a stirred solution of 6-fluoro-l-methyl-4-oxo-7-piperazin-l-yl-4H-[l,3]thiazeto[3,2-α]quinoline-3- carboxylic acid (50 g, 0.143 mole and potassium bicarbonate (15.8 g, 0.1578 mole) in N,N-dimethylformamide (200 ml). The resulting mixture was stirred at 25° to 28°C for 3 to 4 hours. After the completion of the reaction, the reaction mixture was poured into water (1250 ml). The solid obtained was filtered, washed with water (100 ml), and subsequently dissolved in a mixture of chloroform: methanol (7:3; 1250 ml). The lower organic layer was separated and water (500 ml) was added to the organic layer. A dilute aqueous solution of hydrochloric acid was added to the biphasic reaction mixture to adjust pΗ to 0.8 to 1.0. The reaction mixture was stirred for 15 minutes, allowed to settle and the upper aqueous layer was separated. The process was repeated twice and the aqueous layers were combined. Activated charcoal (10%) was added to the combined aqueous layer and stirred for 30 minutes, filtered and cooled to 20° to 25° C. The pΗ of the reaction mixture was adjusted to 6.5 to 7.0 by adding an aqueous solution of sodium bicarbonate. The solid obtained was extracted with chloroform (375 ml), stirred for 15 minutes and the organic layer was separated. The aqueous layer was further extracted with a mixture of chloroform: methanol (7:3 ratio; 50 ml). The combined organic layer was distilled under vacuum at 35° to 40° C to recover the solvent up to 125 ml. The reaction mass so obtained was stirred for 3 to 4 hours at 28° to 30° C, filtered and washed with chilled chloroform (50 ml). The wet cake obtained was dried at 45° C for 12 hours to obtain the title compound. Step B): The prulifloxacin (30 g) obtained in Step A) was suspended in a mixture of chloroform: ethanol (10:1, v/v, 585 ml: 58.5 ml) and heated to reflux temperature. Activated carbon (3.9 gm) was added to the partially cleared solution and refluxed for 30 minutes, followed by filtration through Celite bed. The bed was further washed with chloroform: ethanol (10:1, v/v, 585 ml: 58.5 ml). The filtrate so obtained was distilled at atmospheric pressure till to partially remove the solvent. The concentrate so obtained was stirred at about 25° C for 1 hour, and filtered. The solid obtained was washed with chloroform: ethanol (39 ml X 2), dried under vacuum at 45° C for 12 hours to obtain the title compound. Yield: 22 g
HPLC Purity: 99%
………………………….
SEE
Studies on pyridonecarboxylic acids. 1. Synthesis and antibacterial evaluation of 7-substituted-6-halo-4-oxo-4H-[1,3]thiazeto[3, 2-a]quinoline-3-carboxylic acids
J Med Chem 1992, 35(25): 4727
http://pubs.acs.org/doi/pdf/10.1021/jm00103a011
![]()
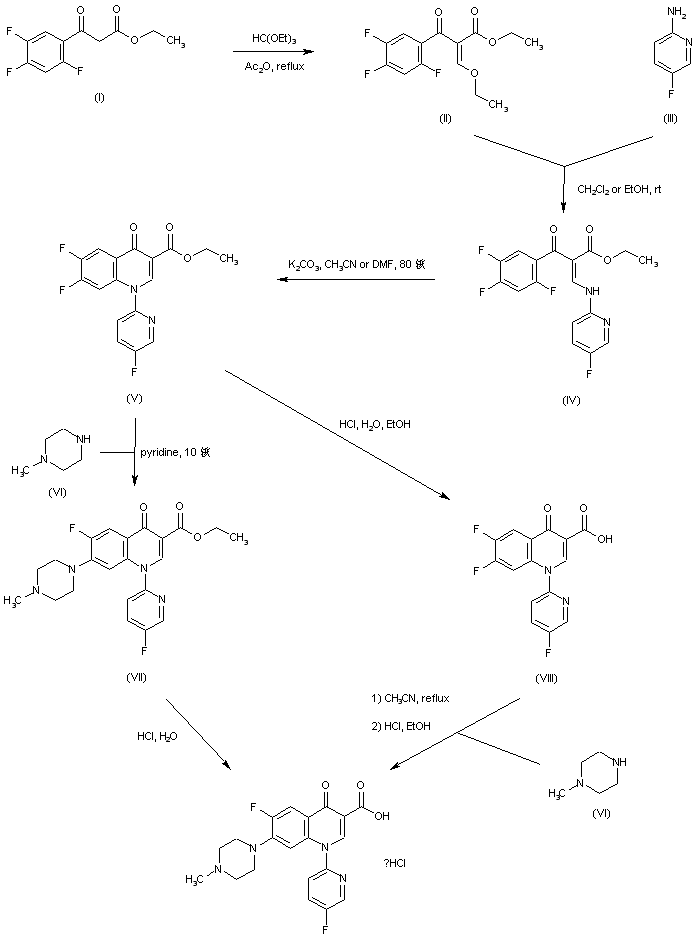
The reaction of 3,4-difluoroaniline (I) with carbon disulfide and triethylamine gives triethylammonium N-(3,4-difluorophenyl)dithiocarbamate (II), which by reaction with ethyl chloroformate and triethylamine in chloroform is converted into 3,4-difluorophenyl isothiocyanate (III). The reaction of (III) with diethyl malonate and KOH in dioxane affords the potassium salt (IV), which is treated with chloromethyl methyl ether in DMF to give the corresponding methoxymethylsulfanyl compound (V). The cyclization of (V) at 240 C in diphenyl ether affords 6,7-difluoro-4-hydroxy-2-(methoxymethylsulfanyl)quinoline-3-carboxylic acid ethyl ester (VI), which by treatment with HCl in ethanol gives the corresponding mercapto compound (VII). The cyclization of (VII) with 1,1-dibromoethane by means of K2CO3 and KI in hot DMF yields 5,6-difluoro-1-methyl-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid ethyl ester (VIII), which is condensed with piperazine (IX) in DMF to afford the corresponding piperazino-derivative (X). The hydrolysis of (X) with KOH in hot tert-butanol gives the corresponding free acid (XI) , which is finally condensed with 4-(bromomethyl)-5-methyl-1,3-dioxol-2-one (XII) by means of KHCO3 in DMF.
………………….
![]()
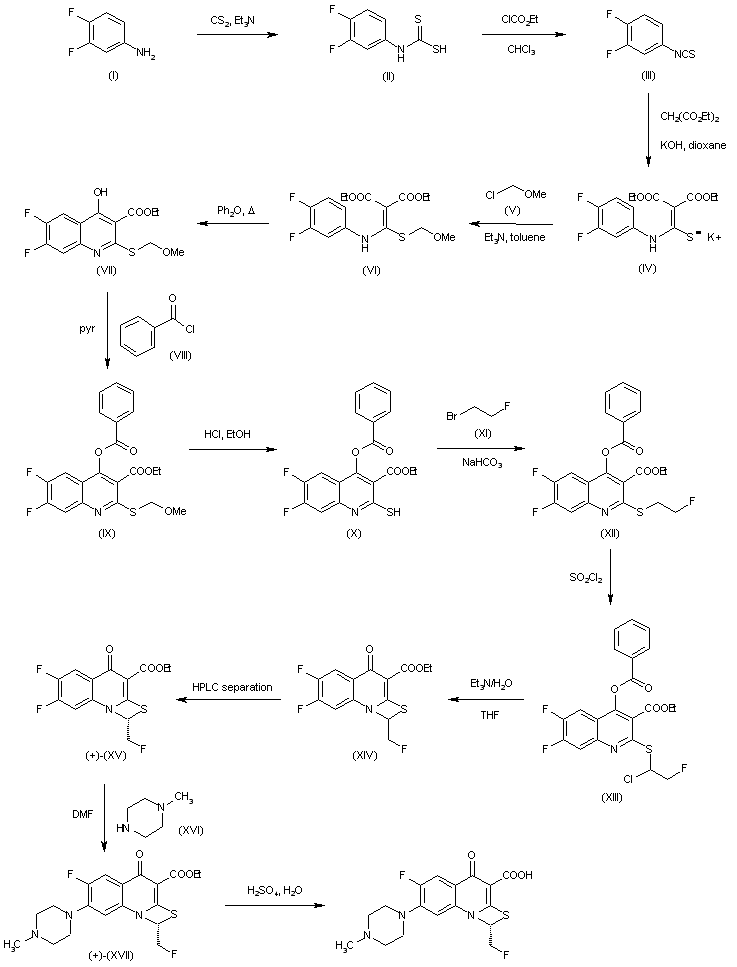
Treatment of 3,4-difluoroaniline (I) with CS2 and Et3N gives triethylammonium dithiocarbamate (II), which reacts with ethyl chloroformate in chloroform to yield (III). Isothiocyanate (III) is converted into the potassium salt (IV) by reaction with diethyl malonate and KOH in dioxane and then transformed into methoxymethyl thioether (VI) by means of reagent (V) and Et3N in toluene. Cyclization of (VI) by heating in diphenyl ether affords quinoline (VII), which then reacts with benzoyl chloride (VIII) in pyridine to furnish (IX). Benzoyloxy derivative (IX) is converted into (X) by means of HCl in EtOH, and its reaction with 1-bromo-2-fluoroethane (XI) and NaHCO3 yields compound (XII). Chlorination of (XII) with SO2Cl2 in hexane provides (XIII), which by simultaneous hydrolysis and intramolecular cyclization by means of Et3N /H2O in THF provides the mixture of isomers (XIV). (+)-(XV) is obtained by HPLC chromatography of (XIV) on a chiral stationary phase. Treatment of (+)-(XV) with 1-methylpiperazine (XVI) in DMF provides ethyl ester (+)-(XVII), which is finally hydrolyzed by means of H2SO4 in H2O.
INTERMEDIATES
![]()
154330-67-3
Ethyl 6,7-difluoro-2-ethylmercapto-4-hydroxyquinoline-3-carboxylate
![]()
154330-68-4
Ethyl 4-acetoxy-6,7-difluoro-2-(ethylthio)quinoline-3-carboxylate
![]()
113046-72-3
Ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate
![]()
113028-17-4
Ethyl 6-fluoro-1-methyl-4-oxo-7-(1-piprazinyl)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate
![]()
112984-60-8
6-Fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid
REFERENCES
- Nelson, Jennifer M.; Chiller, Tom M.; Powers, John H.; Angulo, Frederick J. (2007). “Food Safety: Fluoroquinolone‐Resistant Campylobacter Species and the Withdrawal of Fluoroquinolones from Use in Poultry: A Public Health Success Story”. Clinical Infectious Diseases 44 (7): 977–80. doi:10.1086/512369. PMID 17342653.
- Kawahara S (1998). “[Chemotherapeutic agents under study]“. Nippon Rinsho (in Japanese) 56 (12): 3096–9. PMID 9883617.
- Fritsche, T. R.; Biedenbach, D. J.; Jones, R. N. (2008). “Antimicrobial Activity of Prulifloxacin Tested against a Worldwide Collection of Gastroenteritis-Producing Pathogens, Including Those Causing Traveler’s Diarrhea”. Antimicrobial Agents and Chemotherapy 53 (3): 1221–4. doi:10.1128/AAC.01260-08. PMC 2650572.PMID 19114678.
- Giannarini, Gianluca; Tascini, Carlo; Selli, Cesare (2009). “Prulifloxacin: clinical studies of a broad-spectrum quinolone agent”. Future Microbiology 4 (1): 13–24.doi:10.2217/17460913.4.1.13. PMID 19207096.
- JP patent 1294680, Kise Masahiro; Kitano Masahiko; Ozaki Masakuni; Kazuno Kenji; Matsuda Masato; Shirahase Ichiro; Segawa Jun, “Quinolinecarboxylic Acid Derivative”, issued November 28, 1989
- Prulifloxacin. Drugfuture.com. Retrieved on 2010-11-03.
- Anonymous (2002). “Prulifloxacin ['Quisnon'; Nippon Shinyaku] has been approved in Japan”. Inpharma 1 (1362): 22.
- Research and Development Department of Angelini. Angelinipharma.com. Retrieved on 2010-11-03.
- Nippon Shinyaku, Annual Report 2007
- “Prulifloxacin. NAD-441A, NM 441, Quisnon”. Drugs in R&D 3 (6): 426–30. 2002.PMID 12516950.
- Annual Report 2008, p. 34
Segawa,J,Mashiko kitano, Kenji Kazuno et al, Studies on Pyridonecarboxylic acids,1.Sythesis and antibacterial Evaluation of 7-substituted-6-halo-4-oxo-4H-[1,3]thiazeto [3,2-]quionoline- 3-caroboxylic acids[J].J Med Chem. 1992,35(25):4727-4738.
Masato Matsuoka, Jun Segawa, Yoshihiko.et al, Studies on Pyridone Carb oxylic acids. V.A Practial synthesis of Ethyl 6,7–Difuoro-1-methyl-4-oxo-[1,3] Thiazeto [3,2-a]quinoline-3- Caroboxylate a key intermediate for the new tricyclic quinolone, prulifloxacin (NM441) and Versatile new syntheses of the 2-thioquinoline Skeleton[J].J Heterocyclic Chem.1997,34,1773-1779.
|
|
3-13-1996
|
Sustained release capsule
|
|
|
10-11-1995
|
Method of manufacturing solid dispersion
|
|
|
2-5-1992
|
7(4-(5 METHYL-2-OXO-1,3-DIOXALEN-4-YL)METHYL 1-PIPERZINYL)-4-OXO-4H-(1,3)THIAZETO(3,2-A)QUINOLINE-3-CARBOXYLIC ACIDS
|
|
|
6-31-2011
|
PHARMACEUTICAL COMPOSITION
|
|
|
2-11-2011
|
PROCESS FOR THE PREPARATION OF PURE PRULIFLOXACIN
|
|
|
8-6-2010
|
PROCESS FOR PREPARATION OF PRULIFLOXACIN USING NOVEL INTERMEDIATES
|
|
|
5-7-2010
|
PROCESS FOR THE PREPARATION OF CRYSTALS OF PRULIFLOXACIN
|
|
|
4-9-2010
|
COMPOSITION COMPRISING AN ANTIBIOTIC AND A CORTICOSTEROID
|
|
|
12-11-2009
|
Compounds and Methods for modulating the Silencing of a Polynucleotide of Interest
|
|
|
8-24-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-29-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
7-15-2005
|
Pharmaceutical composition
|
|
|
2-6-2004
|
Medicinal composition
|
| WO2008059512A1 |
Nov 17, 2006 |
May 22, 2008 |
Hetero Drugs Ltd |
Process for preparation of prulifloxacin using novel intermediates |
| WO2008111016A1 |
Mar 14, 2008 |
Sep 18, 2008 |
Ranbaxy Lab Ltd |
Process for the preparation of pure prulifloxacin |
| WO2008111018A2 |
Mar 14, 2008 |
Sep 18, 2008 |
Ranbaxy Lab Ltd |
Process for the preparation of crystals of prulifloxacin |
| WO2010084508A2 |
Dec 10, 2009 |
Jul 29, 2010 |
Elder Pharmaceuticals Ltd. |
Process for the preparation of type i, type ii and type iii crystalline prulifloxacin |
| EP0315828A1 * |
Oct 26, 1988 |
May 17, 1989 |
Nippon Shinyaku Company, Limited |
Quinolinecarboxylic acid derivatives |
| EP1626051A1 |
Apr 28, 2004 |
Feb 15, 2006 |
Nippon Shinyaku Co., Ltd. |
Crystals of quinolinecarboxylic acid derivative solvate |
| US5086049 |
Apr 8, 1991 |
Feb 4, 1992 |
Nipponshinyaku Co., Ltd. |
7[4-(5 methyl-2-oxo-1,3-dioxalen-4-yl)methyl 1-piperzinyl]-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acids |
| US20070149540 |
Apr 28, 2004 |
Jun 28, 2007 |
Nippon Shinyaky Co., Ltd. |
Crystals of quinolinecarboxylic acid derivative solvate |
EXTRA INFO
http://www.google.com/patents/EP2524922A1?cl=en
-
formula 1 is S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl]-4-oxo-4H-[1,3]thiazet o[3,2-α]quinoline-3-carboxylic acid (levo-prulifloxacin for short); its stereo configuration is S configuration; it has optical property of levorotatory polarized light:
-
![Figure imgb0001]()
S-(-) ulifloxacin (as shown in formula 2 below) as raw material and the compound as shown in the following formula 3 are reacted in organic solvent in the presence of alkaline material. The reaction formula is shown below:
![Figure imgb0002]()
-
Accordingly, R-prulifloxacin can be prepared from R-(+)-ulifloxacin and the compound of formula 3 by the method described hereinbefore.
-
[0022]
The reaction formula is depicted below:
-
S-prulifloxacin prepared in accordance with the present invention is determined to be laevorotatory by optical rotation measurement, so it is S-(-)-prulifloxacin. R-prulifloxacin prepared in accordance with the present invention is determined to be dextrorotatory by optical rotation measurement, so it is R-(+)-prulifloxacin.
-
The present invention studied the absorption features of S-(-)-prulifloxacin and R-(+)-prulifloxacin on circular polarized light by circular dichroism spectroscopy. The two spectrograms are mirror images of each other, which proves that S-(-)-prulifloxacin and R-(+)-prulifloxacin are enantiomer of each other.
-
Comparing the circular dichroism spectrogram as depicted in figure 4 with the circular dichroism spectrogram of analogue of the similar structure with known absolute configuration as disclosed in the publication Chem. Pharm. Bull. 47(12) 1765-1773 (1999), it is found that (-)-prulifloxacin has similar Cotton effect to the two analogues reported in the publication, ethyl S-(-)-6,7-difluoro-1-methyl-4-oxo-4H-[1,3] thiazeto[3,2-α]quinoline-3-carboxylate and ethyl S-(-)-6, 7-difluoro-1-fluoromethyl-4-oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylate; so does (+)-prulifloxacin. The results also verify on the other hand that the absolute configuration of levo-prulifloxacin of the present invention is S type while the absolute configuration of dextro-prulifloxacin is R type.
-
The compound of the present invention and physiologically acceptable acid can be prepared to salts: dissolving or suspending S-(-)-prulifloxacin in solvent such as chloroform, DMF and the like; adding into acid or acid solution (for example, hydrochloric acid or hydrogen chloride-methanol solution and the like) while stirring; precipitating and filtering to obtain solid salt from the solvent solution, or alternatively removing solvent from the salt solution directly by concentration, spray drying and the like to obtain the salt of S-(-)-prulifloxacin. The obtained solid may be further recrystallized.
Example 1 Preparation of (S)-(-)-uliflourxacin
-
105 g of racemic uliflourxacin was dissolved in 1,500 mL of dimethyl sulfoxide. 27 g of D-tartaric acid was dissolved in 405 mL of dimethyl sulfoxide dropwise while stirring. After stirring at room temperature for 20 hours, the precipitate was filtrated. The collected solid was dried under vacuum to obtain 86 g solid, which was recrystallized in dimethyl sulfoxide to obtain 37 g of levoulifloxacin-D-tartrate, with C49.08%, H5.06%, N9.50%, S7.44% shown by elemental analysis (molecular formula: C16H16FN3O3S·1/2C4H6O6·H2O, calculated values: C48.86%, H4.78, N9.50%, S7.25%). Said salt was added into water to obtain a suspension, and the pH value was adjusted to 7-8 with 2% NaOH aqueous solution while stirring. After precipitation, filtration, and drying, 24.5 g of (S)-uliflourxacin was obtained, having a chemical name (S)-(-)-6-fluoro-1-methyl-4-oxo-(1-piperazinyl)-1H,4H-[1,3]thiazeto [3,2-α]quinoline-3-carboxylic acid.
-
Specific rotation [α]20 D= -133° (c=0.5, 0.1 mol/L methanesulfonic acid); 1H-NMR (DMSO-d6) δ2.11 (3H, d, j=6.2 Hz), 2.87 (4H, m), 3.19 (4H, m), 6.40 (1H, q, j=6.2 Hz), 6.89 (1H, d, j=7.4Hz), 7.79 (1H, d, j=13.9Hz), optical purity e.e. 96%.
Example 2 Preparation of (R)-(+)-uliflourxacin
-
105 g of racemic uliflourxacin was dissolved in 1,500 mL of DMSO. 27 g of L-tartaric acid was dissolved in 405 mL dimethyl sulfoxide dropwise while stirring to allow that the solution became turbid and the precipitation occurred. The solution was stirred at room temperature for 20 hours and then filtered. The collected solid was dried under vacuum to obtain 82 g solid which was recrystallized in dimethyl sulfoxide to obtain 34 g of dextrouliflourxacin-L-tartarte. Said salt was added into water to obtain a suspension, and the pH value was adjusted to 7-8 with 2% NaOH aqueous solution while stirring. After filtration and drying, 22 g of (R)-uliflourxacin was obtained, having a chemical name (R)-(+)-6-fluoro-1-methyl-4-oxo-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline -3-carboxylic acid.
-
Specific rotation [α]20 D= +132.4° (c=0.5, 0.1 mol/L methanesulfonic acid), optical purity e.e. 96%.
Example 3 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin prepared in Example 1, 2.02 g (0.02 mol) of triethylamine and 20 ml of dimethylformamide (hereinafter referred to as DMF) were mixed and stirred. After the solution was cooled to -5∼5 °C, 0.012 mol of 4-bromomethyl-5-methyl-1,3-dioxolen-2-one (hereinafter referred to as DMDO-Br) in DMF (5 ml) solution was added thereinto, followed by stirring at -5∼5 °C for 3 hours. The reaction solution was poured into 100 ml of ice water, stirred for 30 minutes, and then filtered. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.9 g of S-(-)-prulifloxacin was obtained, having a chemical name: S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl ]-4-oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylic acid, with a purity of 98% and a yield rate of 63%. Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid)
Example 4 Preparation of R-(+)-prulifloxacin
-
R-(+)-prulifloxacin prepared in Example 2 was used as raw material to prepare 2.7 g of target product R-(+)-prulifloxacin in accordance with the method as described in Example 3, with a yield rate of 60.7% and a purity of 98%. Specific rotation [α]20 D= +108° (c=0.5, 0.1 mol/L methanesulfonic acid).
-
Comparing the circular dichroism spectrogram as depicted in Figure 4 with the circular dichroism spectrogram of analogue of the similar structure with known absolute configuration as disclosed in the publication Chem. Pharm. Bull. 47(12) 1765-1773 (1999), it was found that (-)-prulifloxacin has similar Cotton effect with the two analogues reported in the publication, ethyl S-(-)-6,7-difluoro-1-methyl-4-oxo-4H-[1,3] thiazeto[3,2-α]quinoline-3-carboxylate and ethyl S-(-)-6, 7-difluoro-1-fluoromethyl-4-oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylate; so does (+)-prulifloxacin. The results also verify on the other hand that the absolute configuration of levo-prulifloxacin of the present invention is S type while the absolute configuration of dextro-prulifloxacin is R type.
-
Conclusion: The absolute configuration of the sample prepared in Example 3 is S configuration, as shown in the formula below:
Example 5 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin, 1.2 g (0.012 mol) of anhydrous potassium bicarbonate and 20 ml of dimethylsulfoxide were mixed and stirred. 0.012 mol of DMDO-Br in DMSO (5 mL) solution was added dropwise at -20 °C. Stirring proceeded at -20 °C for 3 hours. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.5 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 54%.
Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid)
Example 6 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin, 1.04 g (0.008 mol) of N,N-diisopropylethylamine and 20 mL of N,N-dimethylformamide (DMF) was mixed and stirred, 0.008 mol of DMDO-Br in DMF (5 mL) solution was added thereinto. The solution was heating to 60 °C and reacted for 15 minutes. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.0 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 43%.
Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid)
Example 7 Preparation of S-(-)-prulifloxacin
-
10 g (0.029 mol) of S-(-)-uliflourxacin, 30 ml of N,N-dimethylacetylamide and 14.7 g (0.145mol) of triethylamine was mixed and cooled to 5~10 °C. 8.5 g (0.03 mol) 4-(p-toluenesulfonic acid-1-methyl ester)-5-methyl-1,3-dioxolen-2-one in 25 ml of N,N-dimethylacetylamide solution was added dropwise while stirring. After addition, the solution was reacted at room temperature for 10 hours. The reaction solution was poured into 200 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 7.46 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 57%. Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid).
Example 8 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin, 0.79 g (0.05 mol) of potassium carbonate and 20 ml of dimethylformamide (DMF) was mixed and stirred. 0.012 mol of DMDO-Br in DMF (5ml) solution was added at -10 °C. At the same temperature, the solution was reacted for 2 hours. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.2 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 48%. Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid).
Example 9 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin, 0.79 g (0.02 mol) of diisopropylamine and 20 ml of dimethylformamide (DMF) was mixed and stirred. 0.02 mol of DMDO-Br in DMF (5ml) solution was added at 0 °C. At the same temperature, the solution was reacted for 2 hours. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.5 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 54%. Specific rotation [α]20D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid).
Example 10 Preparation of R-(+)-prulifloxacin
-
In accordance with the method as described in Example 5, the raw material R-(+)-prulifloxacin was prepared to 2.5 g of the target product R-(+)-prulifloxacin with a purity of 98% and a yield rate of 54%. Specific rotation [α]20 D= +108° (c=0.5, 0.1 mol/L methanesulfonic acid).
Example 11 Preparation of levo-prulifloxacin hydrochloride
Example 12 Preparation of levo-prulifloxacin mesylate
Example 13 Preparation of levo-prulifloxacin hydrochloride
Filed under:
Uncategorized Tagged:
PRULIFLOXACIN ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

 ACH 702
ACH 702






















































 burdock
burdock dandelion
dandelion



































































































































 Originally posted on
Originally posted on 



























 EXAMPLE 7 The Preparation of the Compound of Formula (I) (17α-Acetoxy-11β-(4-N,N-dimethylaminophenyl)-19-norpregna-4,9-diene-3,20-dione) From the Compound of Formula (VIII) 340 mL of acetic acid (5.92 mol) were added to a well stirred mixture containing 834 mL of trifluoroacetic anhydride (5.92 mol) in 2,300 mL of methylene chloride under argon. After stirring for 30 minutes at room temperature, 51.3 g of p-toluenesulfonic acid (0.26 mol) were added, and the mixture was chilled to 0 methylene chloride solution containing 128.3 g of the compound of formula (VIII) (0.30 mol) were added, and the reaction mixture was stirred at 0 cautious addition of a 4.5N potassium carbonate solution until the pH was in the range of 7.0-7.5. The reaction mixture was diluted with water and extracted with methylene chloride. The methylene chloride extracts were washed with water and brine, combined, and dried over sodium sulfate.
EXAMPLE 7 The Preparation of the Compound of Formula (I) (17α-Acetoxy-11β-(4-N,N-dimethylaminophenyl)-19-norpregna-4,9-diene-3,20-dione) From the Compound of Formula (VIII) 340 mL of acetic acid (5.92 mol) were added to a well stirred mixture containing 834 mL of trifluoroacetic anhydride (5.92 mol) in 2,300 mL of methylene chloride under argon. After stirring for 30 minutes at room temperature, 51.3 g of p-toluenesulfonic acid (0.26 mol) were added, and the mixture was chilled to 0 methylene chloride solution containing 128.3 g of the compound of formula (VIII) (0.30 mol) were added, and the reaction mixture was stirred at 0 cautious addition of a 4.5N potassium carbonate solution until the pH was in the range of 7.0-7.5. The reaction mixture was diluted with water and extracted with methylene chloride. The methylene chloride extracts were washed with water and brine, combined, and dried over sodium sulfate.









![19-Norpregn-9-ene-3,20-dione, 11-[4-(dimethylamino)phenyl]-5,17-dihydroxy-, cyclic 3,20-bis(1,2-ethanediyl acetal), (5α,11β)-](http://www.lanospharma.com/images/19-Norpregn-9-ene-3,20-dione,%2011-%5B4-%28dimethylamino%29phenyl%5D-5,17-dihydroxy-,%20cyclic%203,20-bis%281,2-ethanediyl%20acetal%29,%20%285%CE%B1,11%CE%B2%29-.gif)

![19-Norpregna-4,9-diene-3,20-dione, 17-(acetyloxy)-11-[4-(methylamino)phenyl]-, (11β)-](http://www.lanospharma.com/images/19-Norpregna-4,9-diene-3,20-dione,%2017-%28acetyloxy%29-11-%5B4-%28methylamino%29phenyl%5D-,%20%2811%CE%B2%29-.gif)













 Relugolix (TAK-385)
Relugolix (TAK-385)


 tak 385
tak 385







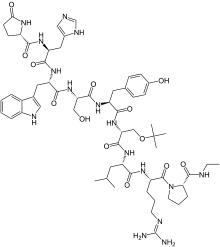 Buserelin
Buserelin
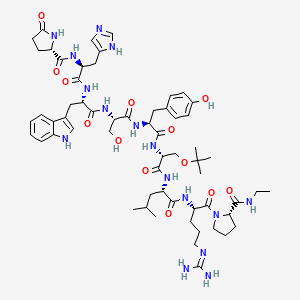 buserelin
buserelin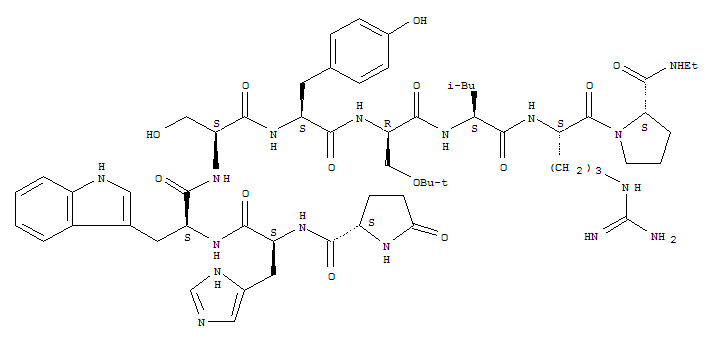








 http://chrom.so/1i2fklC” width=”480″ height=”336″ />
http://chrom.so/1i2fklC” width=”480″ height=”336″ />
























 Dilip sanghvi, sun pharma promoter
Dilip sanghvi, sun pharma promoter