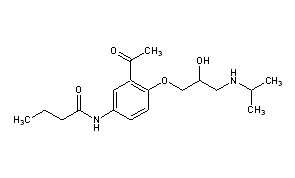
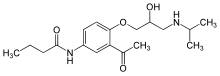
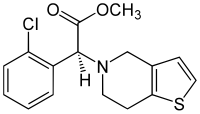
Lifitegrast, SAR 1118
SAR-1118-023
CAS 1025967-78-5
L-Phenylalanine, N-[[2-(6-benzofuranylcarbonyl)-5,7-dichloro-1,2,3,4-tetrahydro-6-isoquinolinyl]carbonyl]-3-(methylsulfonyl)-
![]() INNOVATOR
INNOVATOR
SAR1118 is a white to off-white solid crystallized from methylethylketone. m.p. 154.4oC;
[α]D25=-5.0o(c =1% (w/w) inMeOH); solubility 90 μg/mL in water at 25oC(parent);
FT-IR(KBr): νmax3427, 3302, 3030, 2923, 1727, 1659, 1294, 1140, 826, 764 cm-1;
ESI-MS:m/z615.1[M+1]+, 637.0 [M+Na]+;
1H NMR (300 MHz,d6-DMSO): δ 12.90 (bs, 1H), 9.05 (d,J=6.0Hz,1H), 8.13 (d,J= 1.9 Hz,1H), 7.73 (m, 4H), 7.57 (m, 1H), 7.41 (bs, 1H), 7.05 (d,J= 1.9 Hz,1H),4.78 (bm, 3H),
3.63 (bm, 3H), 3.30 (m, 1H), 3.16 (s, 3H), 3.02 (m, 1H), 2.77 (m, 2H) ppm;
13CNMR (75.5 MHz,d6-DMSO): δ 172.1, 169.6, 163.6, 153.7, 147.8, 140.6, 125.7, 106.9, 53.1,
43.6, 36.4, 26.0 ppm;
Elemental analysis: calcd. for C29H24Cl2N2O7S: C 56.6%, H 3.9%, N 4.6%,S 5.2%, Cl 11.5%; found C 55.1%, H 4.0%, N 4.4%, S 5.2%, Cl 11.2%
![Inline image 4]()
![Inline image 5]()
![Inline image 6]()
SAR 1118 ophthalmic solution from SARcode Bioscience (Brisbane, Calif.) is a first-in-class molecule that inhibits T-cell inflammation by blocking the binding of two key cellular surface proteins (LFA-1 and ICAM-1) that mediate the chronic inflammatory cascade, so it may be able to reduce the inflammation associated with dry-eye disease.
In September, the company initiated enrollment in a Phase III study (OPUS-1). This study will assess the safety and efficacy of SAR 1118 for the treatment of dry-eye disease. Approximately 588 patients will be randomized to receive SAR 1118 5.0% ophthalmic solution or placebo twice daily for 12 weeks. The primary outcome measures include inferior corneal fluorescein staining, vision-related function subscale of the Ocular Surface Disease Index, and safety and tolerability. The company plans to complete the study in the first half of 2012.
The Phase II trial was a randomized, placebo-controlled, multicen-ter trial that included 230 patients with dry eye. In this study, SAR 1118 demonstrated dose-dependent significant improvements in inferior corneal staining over 12 weeks. A statistically significant increase in tear production and improvement in vision-related functions were seen as early as two weeks after initiation of treatment. SAR 1118 was well-tolerated, and no serious ocular adverse events were reported.
Has been found to be an effective inhibitor of Lymphocyte Function- Associated Antigen- 1 (LFA- 1) interactions with the family of Intercellular Adhesion Molecules (ICAM), and has desirable pharmacokinetic properties, including rapid systemic clearance
A growing body of evidence points to a role for inflammation mediated by lymphocyte function-associated antigen-1 (LFA-1) and its ligand intercellular adhesion molecule-1 in the pathogenesis of diabetic macular oedema. This phase 1b clinical trial assessed the safety, tolerability, and pharmacokinetics of topically administered SAR 1118, a novel LFA-1 antagonist, in human subjects
Topical SAR 1118 was safe and well tolerated, and dose-dependent levels of drug were detected in aqueous. However, vitreous levels were below the threshold of detection with the concentrations tested. Further investigation of this medication for posterior segment applications would require intravitreal delivery or chemical modification of the drug.
In a Phase 2 dry eye trial, subjects receiving SAR 1118 demonstrated a reduction in corneal staining, increased tear production, and improved visual-related function as compared to placebo. These data were part of the scientific program of the Association for Research in Vision and Ophthalmology (ARVO) Annual Meeting held in Fort Lauderdale, Florida. SAR 1118 is a first-in-class topically administered small molecule integrin antagonist that inhibits T-cell mediated inflammation, a key component of dry eye disease.
In the randomized, placebo-controlled, multi-center trial, which included 230 subjects with dry eye disease, SAR 1118 demonstrated dose-dependent significant improvements (p<0.05) in inferior corneal staining over 12 weeks. As early as two weeks, a statistically significant(p<0.05) increase in tear production and improvement in visual-related functions (ability to read, drive at night, use a computer, and watch television) were observed, demonstrating early onset of action. Visual-related function was assessed using the Ocular Surface Disease Index (OSDI), a validated instrument designed to measure the severity of dry eye disease and the impact on vision-related function and quality of life. SAR 1118 was safe and well-tolerated with no serious ocular adverse events reported. Most ocular adverse events were transient and related to initial instillation.
“We are encouraged by the clinical effects of SAR 1118 in improving both signs and symptoms of dry eye, which supports the broad anti-inflammatory mechanism of this novel molecule,” commented Charles Semba, MD, Chief Medical Officer of SARcode Corporation. “We are excited to begin Phase 3 development in the later part of 2011, and have discussed appropriate and acceptable endpoints with the regulatory bodies to facilitate a smooth path towards approval.”
“It is well known that dry eye disease can have a deleterious effect on visual function, daily activities, workplace productivity, and quality of life. The potential to impact a patient’s quality of life in as early as 2 weeks could be a major advance in the current dry eye treatment model,” said Quinton Oswald, Chief Executive Officer of SARcode Corporation. “We hope to achieve similar results in our Phase 3 program.”
About Dry Eye Syndrome
Dry eye syndrome is a prevalent and often chronic condition estimated to affect approximately 20 million people in the US. It is among the most common diseases treated by ophthalmologists throughout the world, and has been shown to have a significant impact upon quality of life. Dry eye varies in severity and etiology, and symptoms most commonly manifest as discomfort, visual disturbances, and tear film instability due to decreased quality or quantity of tears. A major contributing factor towards the development of dry eye is inflammation caused by T-cell infiltration, proliferation and inflammatory cytokine production that can lead to reduction in tear film quality and ocular surface damage.
About SAR 1118 – SAR 1118 is a potent novel small molecule lymphocyte function-associated antigen-1 (LFA-1; CD11a/CD18; alphaLbeta2) antagonist under investigation for a broad range of ocular inflammatory conditions including dry eye and diabetic macular edema. LFA-1 is member of the integrin family of adhesion receptors found on the surface of all leukocytes and represents a therapeutic target central to a number of inflammatory stimuli. SAR 1118 has demonstrated potency in blocking LFA-1 binding to its cognate ligand, intercellular adhesion molecule-1 (ICAM-1; CD54), thereby inhibiting cell adhesion, cytokine production, and cellular proliferation in in vitro models.
About SARcode Corporation – SARcode Corporation, founded in 2006, is a venture-backed ophthalmic biopharmaceutical company based in Brisbane, CA. SARcode’s lead development program is a novel class of lymphocyte function-associated antigen-1 (LFA-1) antagonists for the treatment T-cell mediated inflammatory diseases. Institutional investors include Alta Partners and Clarus Venture Partners. For more information, visit http://www.sarcode.com
![Inline image 1]()
![Inline image 1]()
……………………….
http://www.google.com.mx/patents/WO2005044817A1?cl=en
EXAMPLE 14 [0305] This example describes the synthesis of
[0306] which was prepared according to Scheme 9 and the procedure below.
[0307] SCHEME 9
![Figure imgf000097_0003]()
[0308] a) To a solution of 3-carboxylbenzenesulfonyl chloride (3.54 g, 16 mmol) in ethyl acetate (50 mL) at 0 °C was added concentrated ammonia (2.5 mL). The reaction was neutralized with HCl in dioance (20 mL), diluted with ethyl acetate (100 mL), dried with anhydrous sodium sulfate and filtered. Concentration of the filtrate yielded the title compound, which was used without purification. [0309] b) Crude compound 14.1 was dissolved in THF (50 mL), to it was added borane (1.0 M in THF, 50 L) over 20 minute period. After the reaction was stirred at room temperature for 15 hours, the reaction was diluted with brine (20 mL) and water (10 mL), extracted with ethyl acetate (100 mL). The organic extract was dried over anhydrous sodium sulfate and filtered. Concentration of the filtrate yielded the title compound, which was used without further purification. [0310] c) To crude compound 14.2 solution in DCM (100 mL) was added activated 4A molecular sieve powder (8 g), pyridinium dichromate (7.55 g, 20 mmol). After the reaction was stirred at room temperature for 2 hours, the reaction mixture was filtered through silica gel (50 g), rinsed with ethyl acetate. The residue after concentration of the filtrate was purified by silca gel column with 30-50% ethyl acetate in hexane to give compound 14.3 (477mg, 16%, 3 steps). ESI-MS (m/z): (M+H4″) 186. [0311] d) Compound 14.4 was made according to Example 8e except that compound 14.3 was used instead of compound 8.7. MS (ESI4) m/z: 260 (M+H4″). [0312] e) Compound 14 was made according to Example 3g except that compound 14.4 was used instead of compound 3.4. 1H NMR (400 MHz, CD3OD) δ 7.89 (s, 1 H), 7.80 (s, 1 H), 7.75 (m, 2 H), 7.64 (s, 1 H), 7.57(d, 1 H), 7.34 (d, 2 H), 6.93 9s, 1 H), 5.00 (m, 1 H), 3.99 (m, 1 H), 3.73 (m, 1 H), 3.40 (dd, 1 H), 3.12 (dd, 1 H), 2.89 (m, 2 H) ppm; ESI-MS (m/z) 616 (M+H4″). [0313] EXAMPLE 15 [0314] This example describes the synthesis of
![Figure imgf000098_0001]()
which was prepared according to Scheme 10 and the procedure below. [0315] SCHEME 10 rr–λ I BuLi, THF m-CPBA
s ) 2. DMF CH2CI2
15.1 15.2
![Figure imgf000099_0003]()
[0316] a) To a solution of 0.2 mol of furan in 200 mL of dry THF was added 0.2 mol of «-BuLi (1.6 M in hexanes) at -78 °C, the resulting solution was stirred at room temperature for 4 hours. Subsequently, the mixture was cooled to -78 °C and treated with 0.21 mol of dimethyl disulfide, and the mixture was stirred at room temperature overnight, followed by adding 10 mL of saturated aqueous NH C1. The mixture was concentrated at room temperature, and the residue was diluted with 200 mL of saturated aqueous NH4C1 and extracted with ether. The extract was then washed with brine and dried with anhydrous Na2SO . The solvent was removed, and the residue was distilled to collect, the fraction at 135-140 °C/760 mmHg to give compound 15.1 in 55% yield. 1H NMR (400 MHz, CD3C1): δ 7.50 (s, IH), 6.45 (m, IH), 6.39 (s, IH), 2.42 (s, 3H) ppm. [0317] b) To a solution of 0.1 mol of compound 15.1 in 100 mL of dry THF was added 0.1 mol of n- uLi (1.6 M in hexanes) at -78 °C, the resulting solution was stirred at room temperature for 4 hours. Subsequently, the mixture was cooled to -78 °C and treated with 0.12 mol of dry DMF, and the mixture was stirred at room temperature overnight. The reaction was quenched by adding 10 mL of saturated aqueous NH4C1, and the mixture was concentrated. The residue was diluted with 100 mL of brine and extracted with EtOAC. The extract was washed with brine and dried with anhydrous Na2SO4. The solvent was removed and the residue was purified to give the title compound in 65% yield. 1H NMR (400 MHz, CD3C1): δ 9.52 (s, IH), 7.24 (d, J= 3.4 Hz, IH), 6.42 (d, J= 3.4Hz, IH), 2.60 (s, 3H) ppm; ESI-MS (m/z) (M+H4) 143.1. [0318] c) A mixture of 50 mmol of compound 15.2 and 120 mmol of -CPBA in 100 mL of CH2C12 was stirred at room temperature overnight. The mixture was diluted with 150 mL of CH2C12, and the mixture was washed with saturated aqueous NaHCO3 for several times. The solution was then dried with anhydrous Na2SO4 and concentrated. The residue was purified to give compound 15.3 in 70% yield. 1H NMR (400 MHz, CD3C1): δ 9.83 (s, IH), 7.33 (m, 2H), 3.27 (s, 3H) ppm; ESI-MS (m/z): (M+H4″) 175.0.
[0319] d) Compound 15.4 was made according to Example 8e except that compound 15.3 was used instead of 8.7. ESI-MS (m/z): (M+H4″) 248.1. [0320] e) Compound 15 was made according to Example except that compound 15.4 was used instead of 3.4. 1H NMR (400 MHz, CD3OD): δ 7.92 (s, IH), 7.76 (m, IH), 7.67 (s, IH), 7.34 (m, IH), 7.13 (s, IH), 6.69 (s, IH), 6.49 (s, IH), 5.11 (m, IH), 4.73 and 4.88 (m, 2H), 3.76 and 4.02 (m, 2H), 3.46 (m, IH), 3.30 (m, IH), 3.17 (s, 3H), 2.94 (m, 2H) ppm; ESI-MS (m/z): (M+H4) 605.05. [0321]
…………………………………….
US 20110092707
http://www.google.com/patents/US20110092707
Formula I:
has been found to be an effective inhibitor of Lymphocyte Function-Associated Antigen-1 (LFA-1) interactions with the family of Intercellular Adhesion Molecules (ICAM), and has desirable pharmacokinetic properties, including rapid systemic clearance. Improved forms, including crystalline forms, and their uses in treatment of disorders mediated by the interaction of LFA-1 and ICAM are described herein. Novel polymorphs of the compound of Formula I which may afford improved purity, stability, bioavailability and other like characteristics for use in pharmaceutical formulations and methods of use thereof are useful in treating disease.
Methods of Manufacture of the Compound of Formula I
In one embodiment, the compound of Formula I was synthesized as in the following Schemes 1-7. Alternate steps were used in the process as described below. The variants of this overall route yield superior yields, cost of goods and superior chiral purity compared to previously described methods. The final product of this synthesis yields crystalline Form A directly.
![Figure US20110092707A1-20110421-C00009]()
A first alternative protecting strategy produces compound 5, a trityl protected species as shown in Scheme 1. The synthesis begins by reductively aminating 3, 5, dichlorobenzaldehyde, compound 1, with 1-chloro-2-aminoethane and sodium cyanoborohydride in 35% yield. Cyclization of compound 2 using aluminum chloride catalysis and ammonium chloride at 185° C. provided compound 3 in 91% yield. Protection of the free amine of compound 3 as the trityl protected species afforded compound 4 in 89% yield. A carboxylic acid functionality was introduced by treatment of compound 4 with n-butyllithium (nBuLi) and Tetramethylethylenediamine (TMEDA), with subsequent introduction of carbon dioxide, to produce compound 5 in 75% yield.
![Figure US20110092707A1-20110421-C00010]()
Bromophenylalanine was used as the starting material for the right hand portion of the final molecule as shown in Scheme 2. t-Butylcarbamate (Boc) protection of the amino group was accomplished, using sodium bicarbonate (3 equivalents), t-butyl dicarbonate (Boc2O, 1.1 equivalent) in dioxane and water, to obtain compound 7 in 98% yield. A methyl sulfone functionality was introduced by treating the bromo compound 7 with copper iodide (0.4 equivalents), cesium carbonate (0.5 equivalents), L-proline (0.8 equivalents), and the sodium salt of methanesulfinic acid (3.9 equivalents) in dimethylsulfoxide (DMSO) at 95-100° C. for a total of 9 hours, with two further additions of copper iodide (0.2 equivalents) and L-proline (0.4 equivalents) during that period. Compound 8 was isolated in 96% yield. The carboxylic acid of compound 8 was converted to the benzyl ester, compound 9, in 99% yield, using benzyl alcohol (1.1 equivalent), dimethylaminopyridine (DMAP, 0.1 equivalent) and N-(3-dimethylaminopropyl)-N-ethylcarbodiimide (EDC, 1.0 equivalent). The amino group of compound 9 is deprotected by adding a 4N solution of HCl in dioxane to compound 9 at 0° C. in methylene chloride. The HCl salt of the free amino species, compound 10 was isolated in 94% yield.
![Figure US20110092707A1-20110421-C00011]()
Compound 5 was treated with triethylamine (TEA, 5 equivalents) and 2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU, 1.25 equivalents) for 10 minutes in dimethylformamide (DMF), and then compound 10 was added to the solution. After stirring at room temperature for 18 hours, the product, compound 11 was isolated in 70% yield. Removal of the trityl protecting group was accomplished by treating compound 1, with HCl in dioxane (4N, excess) at room temperature for 2 hours, diethyl ether added, and the solid product, compound 12, was isolated by filtration in 95% yield.
![Figure US20110092707A1-20110421-C00012]()
The benzofuranyl carbonyl moiety of the compound of Formula I was prepared using two alternative schemes, Scheme 4 and Scheme 4″. In one embodiment, the benzofuranyl carbonyl moiety was prepared by protecting the hydroxyl group of compound 13 by reacting with tert-butyldimethylsilyl chloride (1.0 equivalents) and triethylamine (TEA, 1.1 equivalents) in acetone, to give compound 14 in 79% yield. A solution of compound 14 in methanol was then treated with sodium borohydride (1.0 equivalent) at room temperature overnight. The reaction was quenched with an addition of acetone, stirred at room temperature for a further 2.5 hours, aqueous HCl (4N) was added with the temperature controlled to below 28C, tetrahydrofuran (THF) was added, and the solution stirred overnight under argon and in the absence of light. The product, compound 15, was isolated quantitatively by extraction into methylene chloride, concentrated at low heat, and used without further purification. The triflate ester, compound 16, was produced in 69% yield from compound 15 by reacting it with N-phenyl-bis(trifluoromethanesulfonimide) (1.0 equivalent) in methylene chloride for 72 hours. Compound 16 in a mixture of DMF, methanol, and triethylamine, was added to a prepared solution of palladium acetate, diphenyl, DMF and methanol in an autoclave. Carbon monoxide was charged into the autoclave to a pressure of 8 bar, and the reaction mixture was heated at 70° C. for 6 hours. After workup, compound 17 was isolated in 91% yield. Lithium hydroxide (4 equivalents) in methanol and water was used to hydrolyze the ester and permit the isolation of compound 18 in 97% yield.
![Figure US20110092707A1-20110421-C00013]()
In one embodiment, the benzofuranyl carbonyl moiety of the compound of Formula I was prepared according to Scheme 4″. By way of an Arbuzov reaction, diethyl 2-(1,3-dioxolan-2-yl)ethylphosphonate, compound 1″, was prepared from 2-(2-bromoethyl)-1,3-dioxolane by the addition of triethyl phosphate. After removal of ethyl bromide through distillation at 210° C. the crude reaction mixture was cooled and then by way of vacuum distillation, compound 1″ was collected as a colorless oil in 94% yield.
In the next step, n-butyllithium (2.15 equivalents) in hexane was cooled to −70° C. and diisopropylamine (2.25 equivalents) was added while keeping the temperature below −60° C. Compound 1″ (1 equivalent) dissolved in tetrahydrofuran (THF) was added over 30 min at −70° C. After 10 min, diethyl carbonate (1.05 equivalents) dissolved in THF was added over 30 min keeping the reaction temperature below −60° C. After stirring for one hour at −60° C., the reaction was allowed to warm to 15° C. and furan-2-carbaldehyde (1.3 equivalents) dissolved in THF was added. After stirring for 20 hrs at room temperature, the reaction was rotary evaporated to dryness to yield ethyl 2-(1,3-dioxolan2-yl)methyl-3-(furan-2-yl)acrylate, compound 5″. Crude compound 5″ was used directly in the next reaction.
The crude compound 5″ (1 equivalent) was dissolved in ethanol and added to a mixture of water and phosphoric acid (85%, 15 equivalents) over 30 min while keeping the temperature below 50° C. After stirring for 20 hrs at room temperature, another 200 ml of phosphoric acid (85%) was added and the mixture was heated to 50° C. for an additional two hrs. After removal of ethanol by rotary evaporation, the material was extracted with toluene, washed with water, dried with sodium sulfate, treated with charcoal, filtered and dried down to an oil. This oil was distilled to afford ethyl benzofuran-6-carboxylate, compound 6″, (bp 111-114.5° C.) which crystallized on standing. Compound 6″ was recovered at 57% yield based on compound 1″.
Compound 6″ (875 mmol) was dissolved in methanol and tetrahydrofuran (THF). Sodium hydroxide (4 M, 3 equivalents) was added and the reaction was stirred overnight. After concentration via rotary evaporation, the aqueous solution was extracted with methyl tert-butyl ether (MTBE), acidified to pH 2 with the addition of hydrochloric acid (HCl) and cooled resulting in fine crystals of benzofuran-6-carboxylic acid, i.e., compound 18. Compound 18 was isolated, washed with water and dried to a final yield of 97% yield.
![Figure US20110092707A1-20110421-C00014]()
The benzofuran carboxylic acid 18 was treated with oxalyl chloride (1.2 equivalents) and a catalytic amount of DMF, stirring for 5.5 hours until a clear solution was obtained. The solvent was removed under reduced pressure and the acid chloride of compound 18 was stored under argon until use, on the next day. The acid chloride, in methylene chloride was added slowly to a methylene chloride solution of the compound of Formula I and diisopropylethylamine (DIPEA) which was cooled to 0-5° C. The reaction was not permitted to rise above 5° C., and after completion of addition, was stirred at 5° C. for a further 0.5 hour. Upon aqueous workup and extraction with methylene chloride, the product, compound 19, was isolated in quantitative yield.
Taking the compound of Formula I directly as the crude reaction product after transfer hydrogenolysis, and reconcentrating down from a solution in methylene chloride, the amorphous form of the compound of Formula I was obtained in 97% purity.
An alternative protection strategy in this synthetic approach is illustrated in Scheme 6.
…………………….
WO 2014018748
http://www.google.com/patents/WO2014018748A1?cl=en
[0040] Methods of Manufacture of the Compound of Formula I
![Figure imgf000009_0001]()
[0041] In one embodiment, the compound of Formula I is synthesized as in the following Schemes 1-7. The final product of this synthesis yields the compound of Formula I as an amorphous solid or as a crystalline form such as Forms A-E, or a pharmaceutically acceptable salt, either directly or indirectly. Variants of this overall route may provide superior yields, cost of goods, and/or superior chiral purity.
[0042] Protecting groups for amino and carboxy groups are known in the art. For example, see Greene, Protective Groups in Organic Synthesis, Wiley Interscience, 1981, and subsequent editions.
[0043] In various embodiments in the subsequent schemes, HATU is used as a reagent in amide- bond forming reactions. Alternatively, HATU is not used. In various embodiments, at least one amide-bond forming reaction is performed with thionyl chloride as a reagent in place of HATU. In various embodiments, all amide-bond forming reactions are performed with thionyl chloride as a reagent to form acid chlorides.
[0044] Scheme 1
[0045] A first alternative protecting strategy produces compound 5′, a protected species as shown in Scheme 1. The synthesis begins by reductively aminating 3, 5, dichlorobenzaldehyde, compound . Cyclization of compound 2′ provides compound 3′. Protection of the free amine of compound 3′ as a protected species provides compound 4′. A carboxylic acid functionality is introduced by treatment of compound 4′ with introduction of carbon dioxide, to produce compound 5′. In various embodiments, the protecting group of compound 4′ is a benzofuranyl carbonyl moiety derived from compound 18′.
[0046] In various embodiments, upon scaleup to multikilogram and larger scale reactions, treatment of compound 4′ with strong base (such as n-butyllithium (nBuLi) to generate a lithio species, or lithium diisopropyl amide (LDA) to generate the lithio species) is performed in flow mode rather than batchwise reaction due to instability of lithio species except at cold temperatures. Flow rates and residence times may be adjusted to maximize yield.
[0047] Scheme IB
5′ 4″”
[0048] In various embodiments, 6-hydroxy-l, 2,3, 4-tetrahydro-isoquino line (Compound 3″) is used as a starting material for Compound 5′. The starting material is chlorinated (x2) for example, with N-chlorosuccinimide. In various embodiments, the chlorination is performed in the presence of a sulfonic acid. In various embodiments, the sulfonic acid is selected from p- toluenesulfonic acid and methanesulfonic acid. Following protection of the amino group, the hydroxy group is functionalized, for example, as the triflate ester, which is carbonylated to yield the amino-protected methyl ester. Hydrolysis of the methyl ester yields the amino protected carboxylic acid.
[0049] Scheme 2
[0050] In various embodiments, bromophenyl alanine is used as the starting material for a portion of the final molecule as shown in Scheme 2. The starting material is protected with an amino protecting group to allow for introduction of a methyl sulfone functionality in compound 8′. Protecting groups are rearranged by introduction of an orthogonal protecting group for the carboxylic moiety, followed by deprotection of the amino group to provide compound 10′. In various embodiments, expensive or exotic bases are replaced with carbonate base such as potassium carbonate or calcium carbonate as a reagent.
[0051] Scheme 2A
10
[0052] In various embodiments, 3-methylsulfonylbenzaldehyde is converted into the 3- methylsulfonylphenylalanine derivative and functionalized to yield compound 10 as shown above.
[0053] Scheme 3
12′
[0054] Compounds 5′ and 10′ are joined through amide bond formation followed by deprotection of the remaining amino group in the presence of the carboxylic protecting group to yield compound 12′ or a salt thereof, such as the HCL salt.
[0055] Scheme 3
[0056] As an alternative to Scheme 3, compound 10″ is coupled with compound 5′ to yield the bromo compound 12″”, with subsequent introduction of a methyl sulfone functionality in place of bromine at a later step to produce compound 19′. Alternatively, instead of a bromine, compound 10″ includes X, where X is any halide (CI, I, Br, F) or a leaving group such as OTs, OTf, or the like.
[0057] Scheme 4
[0058] The benzofuranyl carbonyl moiety of the compound of Formula I can be prepared using various alternative schemes. In one embodiment, the benzofuranyl carbonyl moiety is prepared by protecting the hydroxyl group of compound 13′, reducing the carbonyl of compound 13′ to yield the benzofuranyl moiety, followed by carboxylation to yield compound 18′.
[0059] Scheme 4A
[0060] In one embodiment, compound 18′ is prepared from 6-hydroxybenzofuran via the triflate ester and the 6-carboxy methyl ester as intermediates, as shown in Example 4A.
[0061] Schem
[0062] The benzofuran carboxylic acid 18′ is coupled with compound 12′ (or a salt thereof) by amide bond formation to yield protected compound 19′, as shown in Scheme 5. Amide bond formation is known in the art
[0063] Schem
[0064] As an alternative to Schemes 3-5, compounds 18′ and 5″ may be coupled through amide bond formation followed by deprotection of the remaining carboxylic group to form compound 12″. Amide bond formation between compound 12″ and 10′ yields compound 19′ with a protected carboxylic group.
[0065] Scheme 5B
[0066] As an alternative to Schemes 1-5, compounds 12″ and 10″ may be coupled through amide bond formation followed by introduction of a methyl sulfone functionality in place of the bromine in converting compound 19″ to compound 19′ (similar to Scheme 2). Alternatively, instead of a bromine, compound 10″ includes X, where X is any halide (CI, I, Br, F) or a leaving group such as OTs, OTf, or the like. Compound 12″ can also be made using the following scheme:
[0067] Scheme 6
[0068] Final deprotection of compound 19′ to yield the compound of Formula I or a salt thereof is accomplished in a variety of ways. In various embodiments, the resulting compound of Formula I is provided in higher optical purity and/or higher overall purity and/or higher overall yield.
EXAMPLES
[00111] Example 1
Scheme El
[00112] Reductively aminating 3,5-dichlorobenzaldehyde, compound 1, with l-chloro-2- aminoethane and sodium cyanoborohydride provided 35% yield of compound 2. Cyclization of compound 2 using aluminum chloride catalysis and ammoniun chloride at 185°C provided compound 3 in 91% yield. Protection of the free amine of compound 3 as the trityl protected species afforded compound 4 in 89%> yield. A carboxylic acid functionality was introduced by treatment of compound 4 with n-butyllithium (nBuLi) and tetramethylethylenediamine (TMEDA), with subsequent introduction of carbon dioxide, to produce trityl protected compound 5 in 75% yield.
[00113] Example 1 A
2″
Scheme El A
[00114] To a glass reactor was charged 3,5-dichlorobenzaldehyde. Absolute ethanol was added to the batch slowly (this addition is mildly exothermic) and agitation started. 2,2- Diethoxyethyl amine (1.03 equiv) was slowly added to the batch, keeping the batch temperature at 20-78 °C. The batch was then heated to 76-78 °C for 2 h. GC-MS analysis indicated reaction completion (starting material < 1%). The batch was cooled to ambient temperature for work-up. The batch was concentrated in vacuo to a residue and azeotroped with heptanes (x2). The residue was cooled and held at 0-5 °C for 12 h to form a suspension. The solids were collected by filtration and the cake was washed with cold (0-5 °C) heptanes, and dried under hot nitrogen (45-50 °C) to afford Compound 2′ as a white solid (94% yield).
[00115] To a glass reactor was charged concentrated 95-98%) sulfuric acid (25.9 equiv).
The batch was heated to 120-125 °C and a solution of Compound 2′ in CH2CI2 was added slowly over 1 h, keeping the batch temperature between 120-125 °C. The batch was then stirred at 120— 125 °C for 6 h. The batch was cooled to < 50 °C. To a glass reactor was charged DI water and the batch temperature was adjusted to 0-5 °C. The reaction mixture was slowly transferred, keeping the batch temperature between 0-50 °C. DI water was used to aid the transfer. To the batch was added Dicalite 4200. The batch was filtered through a pad of Dicalite 4200. To the filtrate was added 50% aqueous sodium hydroxide solution slowly over 3 h, keeping the batch temperature between 0-50 °C to adjust the pH to 12. The resulting suspension was stirred at 45- 50 °C for 2 h and the solids were collected by filtration. The filter cake was slurried in DI water at 30-35 °C for 1 h. The batch was filtered. The cake was washed with heptanes and dried in vacuum oven at 45-50 °C for 22 h to give crude compound 2″ as a tan solid (75% yield), which was further purified by recrystallization.
[00116] To a reactor was added platinum dioxide (0.012 equiv), Compound 2″, and
MeOH (10 vol) and the suspension was stirred at room temperature under argon for 10 minutes. The reaction mixture was inerted with argon three times and then stirred under 125 psi of hydrogen at room temperature for 25 hours. HPLC analysis indicated complete reaction with less than 1% of the starting material remaining. After standing, the supernatant was decanted from the solids (catalyst) by vacuum. To the solids was added methanol and the slurry was mixed under nitrogen. The solids were allowed to settle on the bottom over several hours. The supernatant was decanted from the solids by vacuum. The combined supernatants were filtered through Celite under a blanket of nitrogen and the filter pad was washed with MeOH (x2). The combined filtrate and washes were concentrated to dryness. The residue was slurried in MTBE. The mixture was treated with 3 M HC1 while maintaining the temperature <40 °C resulting in the formation of a heavy precipitate. The mixture was stirred at 35-40 °C for 60 to 90 minutes. The batch was cooled to 0-5 °C, stirred for 60 to 90 minutes and then filtered. The filter cake was washed with cold DI water (x2) followed by a displacement wash with MTBE (x2). The filter cake was dried under reduced pressure to afford Compound 3 Hydrochloride Salt (86% yield). The hydrogenation catalyst can be recovered and re-used.
[00117] Compound 3 and trityl chloride were added to the reaction flask. DCM (10 vol) was added to the reactor and agitation was started to form slurry. The reaction mixture was cooled to 10-15 °C. N,N-Diisopropylethylamine (2.5 equiv) was slowly added to the reaction mixture, maintaining the temperature at 15-25 °C during the addition. Once addition was complete, the batch was stirred at 15 to 25 °C for a minimum of 60 minutes. The reaction was assayed by HPLC by diluting a sample with acetonitrile and then injecting it on the HPLC. The first assay after 30 minutes indicated that the reaction was complete with <1% of starting material observed by HPLC analysis. The reaction mixture was diluted with DI water (5 vol). The reaction mixture was stirred for 5 minutes after which it was transferred into a separation funnel and the phases were allowed to separate. The DCM layer was washed with DI water (5 vol) by stirring for 5 minutes and then allowing the phases to separate. The DCM layer was washed with brine (5 vol) by stirring for 5 minutes and then allowing the phases to separate. The DCM layer was dried over magnesium sulfate, filtered and the filter cake was washed with DCM (x2). The combined filtrate and washes were concentrated to a residue that was azeotroped with EtOAc (x2). The residue was suspended in EtOAc and stirred for 1 hour in a 40 °C water bath. The resulting slurry was cooled to 0-5 °C for 1 hour and then filtered. The filter cake was washed twice with EtOAc and then dried under reduced pressure to afford Compound 4.
[00118] Exam le IB
21 4″
[00119] To 1, 2,3, 4-tetrahydro-6-hydroxy-isoqino line in acetonitrile was added p- toluenesulfonic acid and N-chlorosuccinimide. The suspension was cooled to ambient temperature, and the product isolated by filtration for a yield of approximately 61% with purity greater than 95%. The isolated TsOH salt was recrystallized until purity was greater than 99.7%. To one equivalent of the TsOH salt suspended in methanol was added 2M sodium carbonate (0.55 eq.) and 1.2 eq. of Boc anhydride. The suspension was stirred at room temperature overnight. The reaction was monitored by HPLC. Upon completion, the mixture was cooled to below 10 °C, water was added, and the Boc-protected dichloro compound was isolated by filtraton. The product was washed and dried at 40 °C for a yield of 95% and purity of >97%. The Boc-protected dichloro compound was suspended in dichloromethane (10 volumes) and pyridine (5 volumes) was added. The mixture was cooled to below 2 °C, and triflic anhydride (1.25 eq) was added. The mixture was stirred at 0-2 °C for 10 minutes, and then poured into 10 volumes of 6%) aqueous sodium hydrogen carbonate solution. After washing with dichloromethane, the organic phases were combined and dried over magnesium sulphate. Following purification, the product (Compound 4′) was obtained in 90% yield and >98% purity. Compound 4′ was dissolved in dimethylformamide and methanol at room temperature. Diisopropylamine (4 eq) was added. Under CO atmosphere, l,3-bis(diphenylphosphino)propane (0.1 eq) and palladium acetate (0.1 eq) was added. The reaction was heated to refiux, and monitored by HPLC. Upon near completion, the mixture was cooled to ambient temperature. Workup with water, ethyl aceate, and brine yielded Compound 4″, which was used without further purification. Compound 4″ was dissolved in methanol and 2.4 M sodium hydroxide (10 volumes each) and refiuxed. The mixture was cooled to ambient temperature, and toluene was added. Following aqueous workup, the pH was adjusted to 2.3 with 3M hydrochloric acid, and crude product was isolated by filtration in 53% yield with greater than 80% purity.
[00120] Exam le 2
Scheme E2
[00121] t-Butylcarbamate (Boc) protection of the amino group of bromophenyl alanine was accomplished, using sodium bicarbonate (3 equivalents), t-butyl dicarbonate (Boc20, 1.1 equivalent) in dioxane and water, to obtain compound 7 in 98% yield. A methyl sulfone functionality was introduced by treating the bromo compound 7 with copper iodide (0.4 equivalents), cesium carbonate (0.5 equivalents), L-proline (0.8 equivalents), and the sodium salt of methanesulfinic acid (3.9 equivalents) in dimethylsulfoxide (DMSO) at 95-100°C for a total of 9 hours, with two further additions of copper iodide (0.2 equivalents) and L-proline (0.4 equivalents) during that period. Compound 8 was isolated in 96%> yield. The carboxylic acid of compound 8 was converted to the benzyl ester, compound 9, in 99% yield, using benzyl alcohol (1.1 equivalent), dimethylaminopyridine (DMAP, 0.1 equivalent) and N-(3- dimethylaminopropyl)-N-ethylcarbodiimide (EDC, 1.0 equivalent). The amino group of compound 9 is deprotected by adding a 4N solution of HC1 in dioxane to compound 9 at 0°C in methylene chloride. The HCl salt of the free amino species, compound 10 was isolated in 94% yield.
[00122] Example 2 A
[00123] Example 2 was repeated with potassium carbonate in place of cesium carbonate.
[00124] Example 2B
[00125] Boc-protected bromophenylalanine (Compound 7) (100g) was dissolved in
DMSO (400 mL) with stirring and degassing with argon. Sodium methane sulfmate (98g), copper iodide (28.7g), potassium carbonate (40 g) and L-proline (26.75g) were added at 28-30 °C. Reaction was heated to about 87 °C for about 17-19 hours. Reaction was cooled and quenched with crushed ice, stirred for 30-40 minutes, and the pH was adjusted from about 12 to about 3-4 with citric acid (350 g). Quenched reaction mixture was filtered, extracted with dichloromethane x3, washed with ammonium chloride solution, washed with sodium bisulphite solution, and washed with brine. Crude product in dichloromethane was concentrated in vacuo until moisture content was below about 0.5%, and used in next step without further isolation. Crude compound 8 in dichloromethane was charged with benzyl alcohol and DMPA with stirring under nitrogen. Reaction cooled to 0-5 °C. EDC-HCL (1.03 equiv) added with stirring for 30 minutes. Upon completion of reaction by TLC and HPLC, the reaction was quenched with sodium bicarbonate solution, the organic layer was separated, and the aqueous layer was extracted with dichloromethane. The organic layer was washed with citric acid solution, and combined organic layers were washed with brine solution. Dichloromethane was removed at 45- 50 °C, and the concentrate was used for next step without further isolation. The amino group of compound 9 was deprotected by adding a 4N solution of HCl in dioxane to compound 9 at 10- 15°C in methylene chloride. The HCl salt of the free amino species, compound 10 was isolated by filtration from diethyl ether. Isolation of compound 10 was performed through recrystallization using a dimethylformamide/dichloromethane solvent system.
[00126] Example 3
Scheme E3
[00127] Compound 5 was treated with triethylamine (TEA, 5 equivalents) and 2-(7-Aza- lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU, 1.25 equivalents) for 10 minutes in dimethylformamide (DMF), and then compound 10 was added to the solution. After stirring at room temperature for 18 hours, the product, compound 11 was isolated in 70% yield. Removal of the trityl protecting group was accomplished by treating compound 11, with HC1 in dioxane (4 N, excess) at room temperature for 2 hours, diethyl ether added, and the solid product, compound 12, was isolated by filtration in 95% yield. The compound 12 exists in both amorphous and crystalline form and can be isolated in either form.
[00128] Example 3 A
[00129] Compound 5 was dissolved in isopropyl acetate and cooled to 20 to 25 °C.
Thionyl chloride was added, with cooling to 10 to 15 °C, and N-methylmorpholine was added slowly. The reaction was monitored by HPLC. Compound 10, water, and isopropyl acetate were stirred at 15 to 20°C until a solution was achieved. N-methylmorpholine was added followed by addition of the Compound 5 reaction mixture (acid chloride of Compound 5). The reaction was monitored by HPLC. Upon completion, the biphasic layers were allowed to settle, and the aqueous layer was removed. The upper organic layer was extracted with water, and the remaining organic layer was distilled under vacuum. Dioxane and IpAc were added with further distillation. Once dry, 4N anhydrous HC1 in dioxane was added. The mixture was stirred at 20 to 25°C for 12 hours, and checked for complete deprotection by HPLC. Once complete, the thick slurry was filtered, washed with IP Ac and dried under vacuum at 45 to 55°C. Yield of Compound 12 was 88%.
[00130] Example 4
[00131] The benzofuranyl carbonyl moiety of the compound of Formula I was prepared using various schemes, (Schemes E4, E4A, and E4B).
15
Phenyl-bis-triflate
18 ‘
Scheme E4
[00132] The benzofuranyl carbonyl moiety was prepared by protecting the hydroxyl group of compound 13 by reacting with tert-butyldimethylsilyl chloride (1.0 equivalents) and triethylamine (TEA, 1.1 equivalents) in acetone, to give compound 14 in 79% yield. A solution of compound 14 in methanol was then treated with sodium borohydride (1.0 equivalent) at room temperature overnight. The reaction was quenched with an addition of acetone, stirred at room temperature for a further 2.5 hours, aqueous HCl (4N) was added with the temperature controlled to below 28 °C, tetrahydrofuran (THF) was added, and the solution stirred overnight under argon and in the absence of light. The product, compound 15, was isolated quantitatively by extraction into methylene chloride, concentrated at low heat, and used without further purification. The triflate ester, compound 16, was produced in 69% yield from compound 15 by reacting it with N- phenyl-bis(trifluoromethanesulfonimide) (1.0 equivalent) in methylene chloride for 72 hours. Compound 16 in a mixture of DMF, methanol, and triethylamine, was added to a prepared solution of palladium acetate, l,3-Bis(diphenylphosphino)propane (dppp), DMF and methanol in an autoclave. Carbon monoxide was charged into the autoclave to a pressure of 8 bar, and the reaction mixture was heated at 70 °C for 6 hours. After workup, compound 17 was isolated in 91% yield. Lithium hydroxide (4 equivalents) in methanol and water was used to hydro lyze the ester and permit the isolation of compound 18′ in 97% yield.
[00133] Example 4A
[00134] Example 4 was repeated with triflic anhydride and sodium hydroxide as reagents for the ester hydrolysis.
[00135] Compound 15 (6-Hydroxybenzofuran) was stirred in dichloromethane and diisopropylethylamine. Triflic anhydride (1.2 eq.) was added, keeping the temperature below 20C. The reaction was monitored by HPLC. The reaction was quenched with methanol, solvent was removed with vacuum, and the crude residue of Compound 16 was used without further purification. Compound 16 as crude residue was dissolved in 4 volumes of dimethylformamide and 2 volumes methanol. To the solution was added 0.02 eq. of palladium acetate, 0.02 eq. of dppp, and CO under pressure. The reaction was monitored by HPLC. Following workup, Compound 17 was isolated as a crude oily residue without further purification. The residue of compound 17 was dissolved in methanol (5 volumes) and 1 volume of sodium hydroxide (27.65%) was added. The mixture was heated to 40C until full conversion of HPLC. The mixture was cooled to ambient temperature and 3 volumes of water were added. The pH was adjusted to about 2 with 3M hydrochloric acid. The suspension was filtered, washed with water, and dried to give Compound 18′ in about 75% overall yield with purity >99.5%.
[00136] Example 4B
Scheme E4B [00137] Diethyl 2-(l,3-dioxolan-2-yl)ethylphosphonate, compound 1″, was prepared from
2-(2-bromoethyl)-l,3-dioxolane by the addition of triethyl phosphate. After removal of ethyl bromide through distillation at 210°C the crude reaction mixture was cooled and then by way of vacuum distillation, compound 1″ was collected as a colorless oil in 94% yield.
[00138] In the next step, n-butyllithium (2.15 equivalents) in hexane was cooled to -70 °C and diisopropylamine (2.25 equivalents) was added while keeping the temperature below -60 °C. Compound 1″ (1 equivalent) dissolved in tetrahydrofuran (THF) was added over 30 min at -70 °C. After 10 min, diethyl carbonate (1.05 equivalents) dissolved in THF was added over 30 min keeping the reaction temperature below -60 °C. After stirring for one hour at -60 °C, the reaction was allowed to warm to 15 °C and furan-2-carbaldehyde (1.3 equivalents) dissolved in THF was added. After stirring for 20 hrs at room temperature, the reaction was rotary evaporated to dryness to yield ethyl 2-((l,3-dioxolan2-yl)methyl-3-(furan-2-yl)acrylate, which was used directly in the next reaction.
[00139] The crude compound (1 equivalent) was dissolved in ethanol and added to a mixture of water and phosphoric acid (85%>, 15 equivalents) over 30 min while keeping the temperature below 50°C. After stirring for 20 hrs at room temperature, another 200 ml of phosphoric acid (85%>) was added and the mixture was heated to 50 °C for an additional two hrs.
After removal of ethanol by rotary evaporation, the material was extacted with toluene, washed with water, dried with sodium sulfate, treated with charcoal, filtered and dried down to an oil. This oil was distilled to afford ethyl benzofuran-6-carboxylate, compound 6″, (bp 111-114.5°C) which crystallized on standing. Compound 6″ was recovered at 57%> yield based on compound
1″.
[00140] Compound 6″ (875 mmol) was dissolved in methanol and tetrahydrofuran (THF).
Sodium hydroxide (4 M, 3 equivalents) was added and the reaction was stirred overnight. After concentration via rotary evaporation, the aqueous solution was extracted with methyl tert-butyl ether (MTBE), acidified to pH 2 with the addition of hydrochloric acid (HC1) and cooled resulting in fine crystals of benzofuran-6-carboxylic acid, i.e., compound 18′. Compound 18′ was isolated, washed with water and dried to a final yield of 97%> yield.
[00141] Example 5
10% Pd/C, HCOOH/NEt3
MeOH/THF 5:1
Form A of Formula I
Scheme E5
[00142] The benzofuran carboxylic acid 18′ was treated with oxalyl chloride (1.2 equivalents) and a catalytic amount of DMF, stirring for 5.5 hours until a clear solution was obtained. The solvent was removed under reduced pressure and the acid chloride of compound 18′ was stored under argon until use, on the next day. The acid chloride, in methylene chloride was added slowly to a methylene chloride solution of the compound of Formula 12 and diisopropylethylamine (DIPEA) which was cooled to 0-5 °C. The reaction was not permitted to rise above 5°C, and after completion of addition, was stirred at 5°C for a further 0.5 hour. Upon aqueous workup and extraction with methylene chloride, the product, compound 19, was isolated in quantitative yield.
[00143] The benzyl ester of compound 19 was removed by transfer hydrogenolysis using
10% palladium on carbon, using formic acid and triethylamine in a 5: 1 mixture of methanol:THF, to produce the compound of Formula I in 95% yield.
[00144] A final step of slurrying in methyl ethylketone (MEK) produced Form A of the compound of Formula I. The product was washed with water to remove residual MEK. Alternatively, the product of the hydrogenolysis step was slurried in acetonitrile to yield Form A of the compound of Formula I.
[00145] Taking the compound of Formula I directly as the crude reaction product after transfer hydrogenolysis, and reconcentrating down from a solution in methylene chloride, the amorphous form of the compound of Formula I was obtained in 97% purity.
[00146] Example 6
[00147] An alternative protection strategy was performed in Scheme E6.
Scheme E6
[00148] Boc-protection was used for the ring nitrogen in the intermediates 21 and 22.
Compound 5 was deprotected with HC1 in dioxane to produce compound 23 in better than 97%> yield. Boc-protection was introduced, using di-tert-butyl dicarbonate (1.1 equivalent), and compound 21 was obtained in better than 95% yield. Compound 10 was coupled with compound 21 to obtain compound 22, using HATU and triethylamine in DMF. The product, compound 22, was obtained in quantitative yield, and greater than 90% purity. Deprotection with HC1 yielded the compound of Formula 12 in 97.4% yield.
[00149] Transfer hydrogeno lysis of compound 19 produced the compound of Formula I with optical purity of 98.5% (S) enantiomer compared to 79-94.5% (S) enantiomer optical purity obtained by hydrolysis of the corresponding methyl ester.
……………………………..
ACS Med. Chem. Lett., 2012, 3 (3), pp 203–206
DOI: 10.1021/ml2002482
LFA-1/ICAM-1 interaction is essential in support of inflammatory and specific T-cell regulated immune responses by mediating cell adhesion, leukocyte extravasation, migration, antigen presentation, formation of immunological synapse, and augmentation of T-cell receptor signaling. The increase of ICAM-1 expression levels in conjunctival epithelial cells and acinar cells was observed in animal models and patients diagnosed with dry eye. Therefore, it has been hypothesized that small molecule LFA-1/ICAM-1 antagonists could be an effective topical treatment for dry eye. In this letter, we describe the discovery of a potent tetrahydroisoquinoline (THIQ)-derived LFA-1/ICAM-1 antagonist (SAR 1118) and its development as an ophthalmic solution for treating dry eye.
http://pubs.acs.org/doi/suppl/10.1021/ml2002482/suppl_file/ml2002482_si_001.pdf
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| US8084047 * |
Jul 23, 2009 |
Dec 27, 2011 |
Sarcode Bioscience Inc. |
Compositions and methods for treatment of eye disorders |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| US8367701 |
Nov 4, 2011 |
Feb 5, 2013 |
Sarcode Bioscience Inc. |
Crystalline pharmaceutical and methods of preparation and use thereof |
| US8592450 |
Feb 16, 2012 |
Nov 26, 2013 |
Sarcode Bioscience Inc. |
Compositions and methods for treatment of eye disorders |
| US8758776 |
Jan 21, 2011 |
Jun 24, 2014 |
Sarcode Bioscience Inc. |
Compositions and methods for treatment |
| US8771715 |
Jan 21, 2011 |
Jul 8, 2014 |
Sarcode Bioscience Inc. |
Compositions and methods for treatment |
| WO2012121659A1 * |
Mar 8, 2012 |
Sep 13, 2012 |
Kat2Biz Ab C/O Interpares Konsult Ab |
Reduction of c-0 bonds by catalytic transfer hydrogenolysis |
| WO2014018748A1 * |
Jul 25, 2013 |
Jan 30, 2014 |
Sarcode Bioscience Inc. |
Lfa-1 inhibitor and polymorph thereof |
Filed under:
Phase2 drugs Tagged:
dry eye,
LFA-1/ICAM-1 antagonist,
lifitegrast,
ophthalmic solution,
phase 2,
SAR 1118,
tetrahydroisoquinoline derivative ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
 O, O–H
O, O–H
















![[1 with combining macron]](http://www.rsc.org/images/entities/char_0031_0304.gif) space groups, respectively. The improved solubility of telmisartan–saccharin (TEL–SAC) (nine-fold) and telmisartan–glutaric acid (two-fold) cocrystals in comparison with the free drug has been demonstrated in solubility experiments in phosphate buffer, pH 7.5. The TEL–SAC cocrystal remained stable in the aqueous medium for 6 hours as confirmed by PXRD. The AUC0–24 of TEL–SAC and TEL–GA was found to be 2-fold and 1.4-fold increased in terms of bioavailability than pure TEL, respectively. The in vivo antihypertensive activity of TEL–SAC in DOCA salt-induced hypertensive rats showed two-fold improved efficacy, while acute toxicity studies revealed no signs of toxicity in rats even at doses of 2000 mg kg−1 of body weight (BW). The new solid phase of telmisartan with saccharin represents a promising and viable opportunity for the manufacture of a drug product with improved therapeutic outcomes.
space groups, respectively. The improved solubility of telmisartan–saccharin (TEL–SAC) (nine-fold) and telmisartan–glutaric acid (two-fold) cocrystals in comparison with the free drug has been demonstrated in solubility experiments in phosphate buffer, pH 7.5. The TEL–SAC cocrystal remained stable in the aqueous medium for 6 hours as confirmed by PXRD. The AUC0–24 of TEL–SAC and TEL–GA was found to be 2-fold and 1.4-fold increased in terms of bioavailability than pure TEL, respectively. The in vivo antihypertensive activity of TEL–SAC in DOCA salt-induced hypertensive rats showed two-fold improved efficacy, while acute toxicity studies revealed no signs of toxicity in rats even at doses of 2000 mg kg−1 of body weight (BW). The new solid phase of telmisartan with saccharin represents a promising and viable opportunity for the manufacture of a drug product with improved therapeutic outcomes.















 Originally posted on
Originally posted on 

























































 in phase 3
in phase 3 























, click to zoom.")







































 A Chiral Benzoquinolizine-2-carboxylic acid Arginine Salt Active against Vancomycin Intermediate Staphylococcus aureus (VISA). Abstract of Papers, 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, September 2003;American Society for Microbiology: Washington, DC, 2003; Poster F-430
A Chiral Benzoquinolizine-2-carboxylic acid Arginine Salt Active against Vancomycin Intermediate Staphylococcus aureus (VISA). Abstract of Papers, 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, September 2003;American Society for Microbiology: Washington, DC, 2003; Poster F-430































































 Originally posted on
Originally posted on