![Lapatinib3Dan.gif]()
![File:Lapatinib.svg]()
LAPATINIB
Title: Lapatinib
CAS Registry Number: 231277-92-2
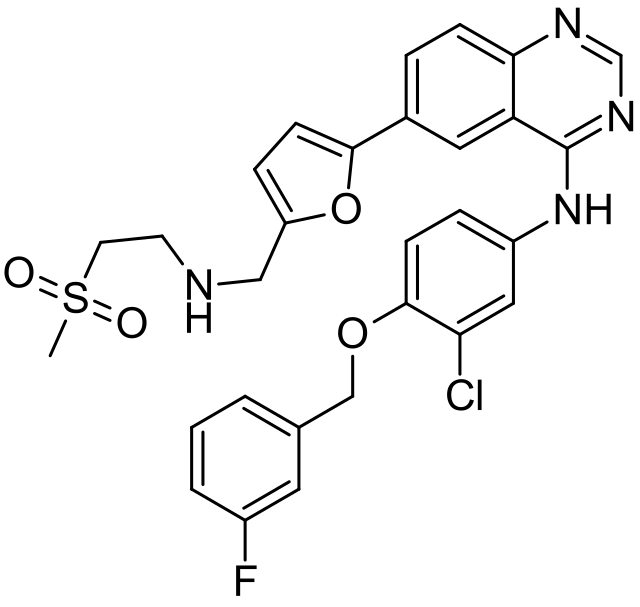
CAS Name: N-[3-Chloro-4-[(3-fluorophenyl)methoxy]phenyl]-6-[5[[[2-(methylsulfonyl)ethyl]amino]methyl]-2-furanyl]-4-quinazolinamine
Trademarks: Tykerb (GSK)
Molecular Formula: C29H26ClFN4O4S
Molecular Weight: 581.06
Percent Composition: C 59.94%, H 4.51%, Cl 6.10%, F 3.27%, N 9.64%, O 11.01%, S 5.52%
![]()
![]()
Lapatinib (INN), used in the form of lapatinib ditosylate, (USAN) (Tykerb/Tyverb, GSK) is an orally active drug for breast cancerand other solid tumours.[1] It is a dual tyrosine kinase inhibitor which interrupts the HER2/neu and epidermal growth factor receptor(EGFR) pathways.[2] It is used in combination therapy for HER2-positive breast cancer. It is used for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpress HER2 (ErbB2).[3]
Status
On March 13, 2007, the U.S. Food and Drug Administration (FDA) approved lapatinib in combination therapy for breast cancer patients already using capecitabine (Xeloda, Roche).[2][3] In January 2010, Tykerb received accelerated approval for the treatment of postmenopausal women with hormone receptor positive metastatic breast cancer that overexpresses the HER2 receptor and for whom hormonal therapy is indicated.[3]
Pharmaceutical company GlaxoSmithKline (GSK) markets the drug under the propriety names Tykerb (mostly US) and Tyverb (mostly Europe).[4] The drug currently has approval for sale and clinical use in the US,[2][4] Australia,[2] Bahrain,[2] Kuwait,[2] Venezuela,[2]Brazil,[5] New Zealand,[5][6] South Korea,[5] Switzerland,[4] Japan, Jordan, the European Union, Lebanon, India and Pakistan.[4]
On the 2nd of August 2013, India’s Intellectual Property Appellate Board revoked the patent for Glaxo’s Tykerb citing its derivative status, while upholding at the same time the original patent granted for Lapatinib.[7]
The drug lapatinib ditosylate is classified as S/NM (a synthetic compound showing competitive inhibition of the natural product) that is naturally derived or inspired substrate (Gordon M. Cragg, Paul G. Grothaus, and David J. Newman, Impact of Natural Products on Developing New Anti-Cancer Agents, Chem. Rev. 2009, 109, 3012–3043)
Breast cancer[
Lapatinib is used as a treatment for women’s breast cancer in treatment naive, ER+/EGFR+/HER2+ breast cancer patients(now often called “triple positive”) and in patients who have HER2-positive advanced breast cancer that has progressed after previous treatment with other chemotherapeutic agents, such as anthracycline, taxane-derived drugs, or trastuzumab (Herceptin, Genentech).
A 2006 GSK-supported randomized clinical trial on female breast cancer previously being treated with those agents (anthracycline, a taxane and trastuzumab) demonstrated that administrating lapatinib in combination with capecitabine delayed the time of further cancer growth compared to regimens that use capecitabine alone. The study also reported that risk of disease progression was reduced by 51%, and that the combination therapy was not associated with increases in toxic side effects.[11] The outcome of this study resulted in a somewhat complex and rather specific initial indication for lapatinib—use only in combination with capecitabine for HER2-positive breast cancer in women whose cancer have progressed following previous chemotherapy with anthracycline, taxanes and trastuzumab.
![]()
………………………………………………………..
US6727256
or
http://www.google.co.in/patents/WO1999035146A1
………………………………………………..
http://www.google.com/patents/EP2550269A1?cl=en
Lapatinib has the structural formula (I) and chemical name N-[3- chloro-4-[(3-fluorophenyl)methoxy]phenyl]-6-[5-[(2-methylsulfonylethylamino)methyl]-2- furyl] quinazolin-4-amine.
BACKGROUND ART
Lapatinib is a tyrosine kinase inhibitor that is used as an orally administered drug as its ditosylate salt to treat certain types of advanced or metastatic breast cancer and other solid tumors. Lapatinib ditosylate was approved by the FDA in 2007 and the EMEA in 2008 and is marketed by GlaxoSmithKline (GSK) under the trade name of Tykerb® in the USA and Tyverb® in Europe.
Lapatinib substance is claimed in US 6,713,485 B2 and US 6,727,256 Bl and lapatinib ditosylate and its crystalline forms are claimed in US 7,157,466 B2. A synthesis of lapatinib that utilises a palladium mediated coupling of a substituted 4-anilino-6-iodo-quinazoline (II) with a 2- (tributylstannyl)furan (Ilia) is disclosed in US 6,727,256 Bl and is also presented in US 7,157,466 B2. In US 7,157,466 B2 a second generation approach was disclosed that utilises a palladium catalysed coupling of a substituted 4-anilino-6-iodo-quinazoline (II) with furan-2-yl-boronic acids (Illb). Following the palladium catalysed coupling reactions utilised in the two synthetic methods of US 6,727,256 Bl and US 7,157,466 B2, only one (US 7,157,466 B2) or two (US 6,727,256 Bl and US 7,157,466 B2) synthetic modification of the structure are utilised before the lapatinib substance is provided (Scheme 1). The EMEA’s COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) has published guidelines titled GUIDELINE ON THE SPECIFICATION LIMITS FOR RESIDUES OF METAL CATALYSTS OR METAL REAGENTS and recommendations are presented for oral exposure to metals, including palladium. For a drug being consumed in quantities not exceeding a 10 g daily dose, a limit of 10 ppm (parts per million) concentration of palladium in the drug substance is recommended. Given this, there is still an unmet need for an alternative synthetic method that can be used for preparation of lapatinib in which the palladium mediated coupling step is performed early in the synthetic route, thereby being capable to provide .
![Figure imgf000004_0001]()
Scheme 1
SUMMARY OF THE INVENTION
There are a number of ways that the levels of a metal, such as palladium, can be controlled in a drug substance through purging of the metal by treatment of the drug substance or its synthetic intermediates or both, including crystallisation, aqueous extraction, filtration through metal absorbent filter aids (Organic Process Research & Development 2005, 9, 198-205), precipitation of the metal from solution, chromatography, and treatment with metal scavenging reagents (Organic Process Research & Development 2003, 7, 733-742). By placing the palladium mediated coupling step downstream in the synthetic route, however, to take advantage of synthetic convergence, the opportunity to reduce the level of palladium in the drug substance is reduced. In contrast, however, by redesigning the synthetic route to move the palladium mediated coupling step upstream, further away from the drug substance, increases the opportunity to control the palladium level in the drug substance. Furthermore, by careful operational design (such as in a precipitation and crystallisation step), the palladium level in the intermediates can be consistently controlled. Given that there is a need, the present invention has addressed these two latter points and utilised them in a novel and efficient process for the manufacture of lapatinib and lapatinib ditosylate.
![Figure imgf000005_0001]()
Scheme 2 – Synthesis of lapatinib and lapatinib ditosylate
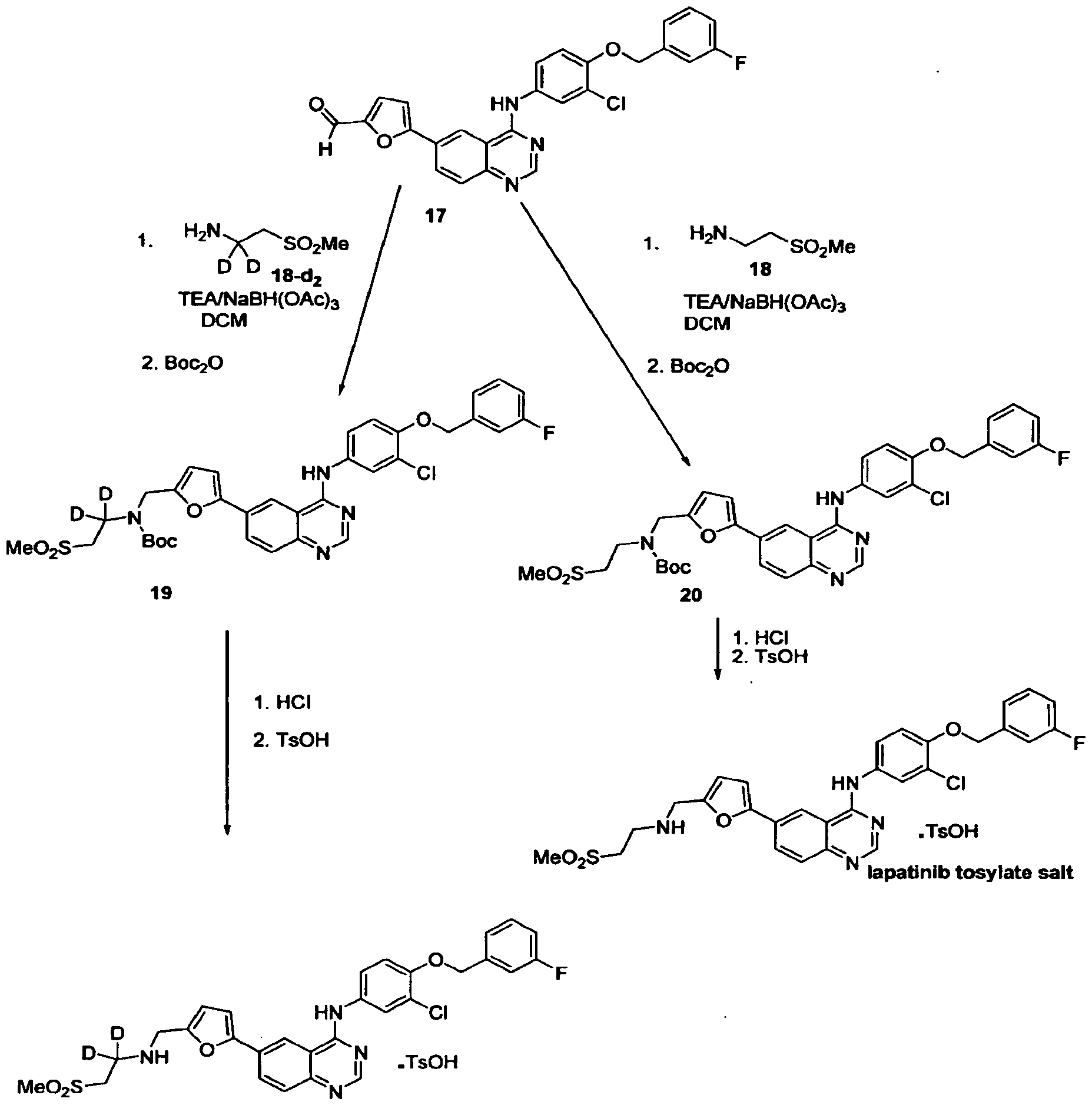
In contrast to the prior art methods disclosure in US 6,727,256 Bl and US 7,157,466 B2, the present invention has performed a transition metal catalysed coupling reaction at the most upstream point in the synthetic route based on the utilization of commercially available starting materials SMla (6-iodoquinazolin-4(3H)-one) and SM2a (5-formylfuran-2-ylboronic acid), or their analogues SMI and SM2, to provide IM1. Thus, in one aspect of the present invention, lapatinib is made from a novel compound (IM1) (Scheme 2).
In another aspect of the present invention, a lapatinib ditosylate monohydrate is prepared by crystallizing lapatinib ditosylate in a mixture of water, DMSO and MeCN.
In another aspect of the present invention, novel compound IM1 is synthesized by the cross- coupling of commercially available SMla and SM2a, or their analogues SMI and SM2, in suitable solvents comprised of an organic solvent and water in the presence of a base and a catalyst formed from a transition metal and a ligand (scheme 3).
X = CI, Br, I, OTf Y = CHO, or CH(OR)2
BZ = B(OH)2, B(OR)2, [BF3]M or BR2
Scheme 3
Example
Example 1: Synthesis of 5-(4-oxo-3,4-dihydroquinazolin-6-yl)furan-2-carbaldehyde (IMl)
IM1
A 5:2 v/v mixture of DMSO and H20 (1400 mL) was degassed for 30 min at ambient temperature using nitrogen. 5-Formylfuran-2-ylboronic acid (SM2a; 26.8 g, 193 mmol) was added dissolved in this mixture. [HP(i-Bu)3] BF4 “ (840 mg, 2.94 mmol) and Pd(OAc)2 (680 mg, 2.94 mmol) was added and the mixture was stirred at ambient temperature under an atmosphere of nitrogen for 20 min. AcOK (18.8 g, 192 mmol) was added into the reactor and was stirred for 20 min at ambient temperature. 6-Iodoquinazolin-4(3 /)-one (SMla; 40 g, 147 mmol) was added and heated to 80±5°C (internal temperature) in an oil bath under nitrogen, Upon completion of the reaction (HPLC), the reaction mixture was hot-filtered, then hot water (400 mL, 80±5°C) was added into the filtrate. This was slowly cooled to 0-15°C (solid started to precipitate at 70°C (internal temperature) and was then filtered. The filter cake was washed with H20 (80 mL), then with MeCN (60 mL), and dried in vacuo at 60+5°C for 6 h to provide 5-(4-oxo-3,4-dihydroquinazolin-6-yl)-furan-2- carbaldehyde (IMl; 34.6 g, 144 mmol) with 99.7 % HPLC purity in 97.6% HPLC yield. XH NMR (300 MHz, de-DMSO): δ 7.47 (d, / = 3.8 Hz, 1H), 7.69 (d, / = 3.8 Hz, 1H), 7.77 (d, / = 8.6 Hz, 1H), 8.17 (s, 1H), 8.27 (dd, / = 8.6, 2.1 Hz, 1H), 8.52 (d, = 2.1 Hz, 1H), 9.66 (s, 1H); 13C NMR (75 MHz, CDC13): δ 110.5, 122, 6, 123.9, 126.0, 127.5, 129.0, 131.4, 147.1, 150.1, 152.7, 157.6, 161.2, 178,8; ESI-MS, Pos: [M+H]+ mJz 241; IR (cm 1): 1713, 1671, 1604,1462; m.p.: 267°C. See Figure 2 for the DSC/TGA of IMl; See Figure 3 for the X-ray powder diffraction pattern of IMl; Residual concentration of palladium: 230 ppm.
Example 2: Synthesis of 5-(4-chloroquinazolin-6-yl)furan-2-carbaldehyde hydrochloride
(IM2a.HCl)
I 1 reflux IM2a.HCI
Over a 1.5 hour period under an atmosphere of N2, SOCb (86.2 g) in MeCN (145 mL) was added dropwise into a mixture, that had been preheated at reflux for 0.5 h, of IM1 (29 g, 0.121 mol), MeCN (435 mL) and DMF (0.88 g) at reflux. The reaction was terminated when less than 2% (HPLC) of IM1 was remaining. If the reaction did not achieve complete reaction, extra SOCI2was added. The mixture was cooled to about 25±5°C (internal temperature), and was then filtered and washed with MeCN (58 mL) to give ca. 55 g of IM2a.HCl (moist with MeCN) with 82A purity by HPLC. IM2a.HCl: ¾ NMR (300 MHz, d6-DMSO): δ 9.68 (s, 1 H), 9.17 (s, 1H), 8.57 (d, / = 2.0 Hz, 1H), 8.46 (dd, J = 8.6, 2.1 Hz, 1H), 8.02 (d, / = 8.6 Hz, 1H), 7.74 (d, = 3.8 Hz, 1H), 7.60 (d, J = 3.8 Hz, 1H). See Figure 5 for the XH NMR spectrum of IM2a.HCl; 13C NMR (75 MHz, d6- DMSO) δ 179.0, 159. 6, 156.4, 152.9, 149.5, 141.0, 132.6, 129.2, 125.9, 123.2, 122.9, 122.7, 111.5;
IM2a.HCl was purified by column chromatography (eluent: ) to give pure IM2a. IM2a: lH NMR (300 MHz, d6-DMSO): δ 7.53 (d, / = 3.3 Hz, 1H), 7.68 (d, J = 3.3 Hz, 1H), 8.02 (d, / = 8.7 Hz, 1H), 8.42 (d, / = 8.4 Hz, 1H), 8.54 (d, / = 2.1 Hz, 1H), 8.90 (s, 1H), 9.64 (s, 1H); 13C NMR (75 MHz, CDCI3): δ 111.5, 122.8, 122.9, 123.7, 125.9, 129.1, 132.5, 142.1 , 149.3, 152.9, 156.6, 159.7, 179.1.
Example 3: Synthesis of 5-(4-(3-chloro-4-(3-fluorobenzyloxy)phenylamino)
- uinazolin-6-yl)furan-2-carbaldehyde hydrochloride (IM3.HC1)
A mixture of IM2a.HCl (moist with MeCN solvent, prepared from 29 g IM1, 0.120 mol) and 3-chloro-4-(3-fiuorobenzyloxy)aniline (SM3; 27.3 g, 0.108 mol) in MeCN (580 mL) was stirred under reflux, until HPLC analysis showed that the reaction was completed (about 2 h). The mixture was cooled to room temperature (25±5°C), filtered, and washed with MeCN (58 mL). A mixture of the moist crude solid IM3 and THF (870 mL) was treated with a 2.0 N aqueous NaOH (348 mL) and stirred for 3-4 h until most of the solid had dissolved. The mixture was filtered through diatomite and was washed with a saturated aqueous solution of NaCl (87 mL). The organic layer was treated with 10% aqueous HCI (174 mL) and stirred for 0.5 h. The resulting solid was filtered, washed with THF (87 mL), and dried in vacuo at 60+5°C for 16 h to give the crude IM3.HC1 (34 g, 0.067 mol, HPLC purity: 99%).
IM3.HC1: :H NMR (300 MHz, d6-DMSO): δ 9.69 (s, 1H), 9.52 (s, 1H), 8.94 (s, 1H), 8.50 (dd, / = 8.8, 1.7 Hz, 1H), 8.01 (d, / = 8.8 Hz, 1 H), 7.97 (d, J =2.5 Hz, 1H), 7.77 (d, / = 3.8 Hz, 1H), 7.73 (dd, = 9.0, 2.5 Hz, 1H), 7.69 (d, / = 3.8 Hz, 1H), 7.49 (td, 7 = 8.0, 6.1 Hz, 1 H), 7.41-7.28 (m, 3H), 7.20 (td, / = 8.4, 2.2 Hz, 1H), 5.31 (s, 2H).
Free base IM3 is obtained by column chromatography (eluting with EtOAc/DCM, 1:4, v/v). IM3 XH NMR (300 MHz, d6-DMSO): δ 5.28 (s, 2H), 7.19 (td, /= 8.7 Hz, 7 = 2.1 Hz 1H), 7.34 (m, 4H), 7.43 (d, 7 = 3.6 Hz , 1H), 7.49 (m, 1H), 7.73 (dd, 7 = 8.7 Hz 7 = 2.7 Hz, 1H), 7.76 (d, 7 = 3.6 Hz, 1H), 7.88 (d, 7 = 9 Hz, 1H), 8.07 (d, 7 = 2.1 Hz, 1H), 8.32 (dd, 7 = 4.43 Hz, 7 = 1.95 Hz, 1H), 8.95 (d, 7 = 1.5 Hz, 1H), 9.68 (s, 1H).
Example 4: Synthesis of N-(3-chloro-4-(3-fluorobenzyloxy)phenyl)-6-(5-((2- (methylsulfonyl)ethylamino)methyl)furan-2-yl)quinazolin-4-amine ditosylate (lapatinib ditosylate)
I
To a suspension of 2-(methylsulfonyl)ethanamine hydrochloride (SM4.HC1; 12.2 g, 76.7 mmol) in THF (600 mL) was added acetic acid (14.1 g, 235 mmol) followed by DIPEA (30.3 g, 235 mmol) were added. After stirred at ambient temperature for 0.5 h, ¾0 (4.2 g, 233 mmol) and IM3.HC1 (30.0 g, HPLC assay >99%, 58.7 mmol) were added. After being stirred at ambient temperature (20°C) for 4 h, sodium triacetoxyborohydride (37.4 g, 176 mmol) was added and the mixture was stirred at ambient temperature (20°C±5°C; external temperature) until HPLC showed the completion of the reaction. A 10% aqueous solution of sodium hydroxide (90 mL) was added and the mixture was stirred for 30 min. The organic phase was washed with 25% aqueous NH4C1 (60 mL), filtered, treated with -TsOH (40.4 g, 135 mmol) and heated to reflux for 2 h. The mixture was cooled to ambient temperature and stirred for 3 h at ambient temperature. The mixture was filtered, and the filter cake was washed twice with THF (120 mL each) and was then dried under vacuum at 70±5°C for 6 h to give 43 g (46.5 mmol) lapatinib ditosylate with 99.4% HPLC purity.
Lapatinib ditosylate [H NMR (300 MHz, d6-DMSO): δ 11.41(s, 2H), 9.33 (s, 3H), 9.04 (d, / = 1.3 Hz, 2H), 8.93 (s, 2H), 8.41 (dd, J =8.8, 1.6 Hz, 2H), 7.91 (d, J = 2.6 Hz, 2H), 7.54-7.41 (m, 9H), 7.37 – 7.27 (m, 6H), 7.25 (d, / = 3.4 Hz, 2H), 7.22 – 7.13 (m, 2H), 7.08 (dd, / = 8.4, 0.6 Hz, 8H), 6.87 ( d, / = 3.5 Hz, 2H), 5.29 (s, 4H), 4.46 (s, 4H), 3.65 – 3.51 (m, 4H), 3.51 – 3.38 (m, 4H), 2.26 (s, 12H).
A solution of lapatinib ditosylate was converted to its free base form, lapatinib, by washing a solution with aqueous NaOH followed by concentration. Lapatinib: XH NMR (300 MHz, d6-DMSO): δ 2.98 (t, / = 6.75 Hz, 1H), 3.04 (s, 1H), 3.29 (t, J = 6.6 Hz, 1H), 3.83 (s, 1H), 5.28 (s, 1H), 6.50 (d, / = 3.0 Hz, 1H), 7.08 (d, / = 3.3 Hz, 1H), 7.20 (m, 1H), 7.33 (m, 4H), 7.48 (m, 1H), 7.76 (m, 1H), 7.80 (d, 7 = 9 Hz, 1H), 8.04 (d, 7 = 2.75 Hz, 1H), 8.17 (dd, / = 8.7 Hz, / = 1.8 Hz, 1H), 8.56 (s, 1H), 8.75 (d, J = 1.8 Hz, 1H).
Example 5a: Purification of lapatinib ditosylate
Lapatinib ditosylate (5.0 g, 5.4 mmol, 96.5% HPLC purity with the maximum individual impurity at 0.8%) was dissolved in DMSO (10 mL) at 70°C (internal temperature). MeCN (10 mL) was added dropwise into the mixture at 70-80°C (internal temperature) and was stirred at this temperature for 1 h. Over a 4 h period the mixture was cooled to room temperature. MeCN (30 mL) was added dropwise, and the mixture was stirred for lh, then filtered and washed with MeCN (10 mL). The filter cake was dried under vacuum at 60°C for 16 h to give 4.0 g lapatinib ditosylate as crystalline Form 1 (as disclosed in US 7,157,466 B2) with 99.6% HPLC purity in 78% HPLC yield.
Example 5b. Purification of lapatinib ditosylate.
Lapatinib ditosylate (3 g, 3.25 mmol, 99.3% HPLC purity was dissolved in DMF (18 mL) at 80°C and stirred for 1 hour. The mixture was hot-filtered. MeCN (18 mL) was added into the filtrate at 80°C. The temperature was cooled to 70°C and crystal precipitated. The mixture was kept at 70°C for 1 h and then 60°C for 1 h. The mixture was further cooled to 0°C and stirred for 2 h. The crystals of lapatinib ditosylate were isolated by filtration and were dried at 40°C under vacuum overnight. Lapatinib ditosylate (2.5 g, 2.70 mmol, 83% yield) with 99.9% HPLC purity was obtained. XRPD analysis (figure 9) indicated that this was Form 2 as disclosed in WO 2009/079541 Al.
Example 6: Preparation of lapatinib ditosylate monohydrate Lapatinib ditosylate (2.0 g, 96.7% HPLC purity, 2.1 mmol) was dissolved in DMSO (5 mL) at 80°C (internal temperature) and the solution was filtered whilst the lapatinib ditosylate was still dissolved. A mixture of MeCN (5 mL, 2.5 P) and water (0.3 mL) was then added dropwise into the filtered solution at 70-80°C (internal temperature). The mixture was cooled at a rate of 10°C/h until 60°C, and was kept at 60°C for 2 h and was then slowly cooled down to 50°C. After being kept at 50°C for 1 h, MeCN (15 mL) was added, and then the mixture was cooled to 20-30°C and stirred at 20-30°C for 2 h. The slurry was filtered, washed with MeCN (6 mL) and the filter cake was dried in vacuo at 60°C for 4 h to give lapatinib ditosylate monohydrate (1.7 g, 99.4A% purity, 1.8 mmol). XRPD analysis (figure 10) indicated that this was the monohydrate crystalline form as disclosed in US 7,157,466 B2.
![]()
…………………………………………………
Beilstein J. Org. Chem. 2013, 9, 2265–2319.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
GlaxoSmithKline’s lapatinib (3.38, Tykerb) is a novel dual kinase inhibitor used in the treatment of solid tumors such as those found in breast cancer and contains a quinazoline core structure. It consists of a 2,5-disubstituted furan ring, which is directly linked to the aminoquinazoline unit (Scheme 41). The quinazoline heterocycle was prepared starting from 5-iodoanthranilic acid (3.72) via initial condensation with formamidine acetate (3.73) followed by chlorination using oxalyl chloride or phosphorous oxychloride [101]. Performing a nucleophilic aromatic substitution on the chloride 3.74 with aniline 3.75renders the extended core of lapatinib. This intermediate (3.76) was then coupled with 5-formyl-2-furanoboronic acid (3.77) using standard Suzuki cross-coupling conditions. Finally, a reductive amination of the pendant aldehyde of3.78 with 2-(methylsulfonyl)ethylamine (3.79) furnishes the desired product lapatinib (Scheme 41).
Synthesis of lapatinib.
get ref from
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
……………………………………………..
Guntrip SB, Lackey KE, Cockerill GS, Carter MC, Smith KJ Bicyclic heteroaromatic compounpds as protein tyrosine kinase inhibitors. EP 1047694; WO 9935146.
![]()
Quinazoline ditosylate salt compounds (US7157466)
A NOVEL PROCESS FOR THE PREPARATION OF Lapatinib AND ITS PHARMACEUTICALLY ACCEPTABLE SALTS ( WO 2010061400)
…………………………………………………….
Fresenius Kabi Oncology Ltd.WO 2013080218
Lahiri, Saswata; Gupta, Nitin; Singh, Hemant Kumar; Handa, Vishal; Sanghani, Sunil
6 JUNE 2013, http://www.google.com/patents/WO2013080218A1?cl=en
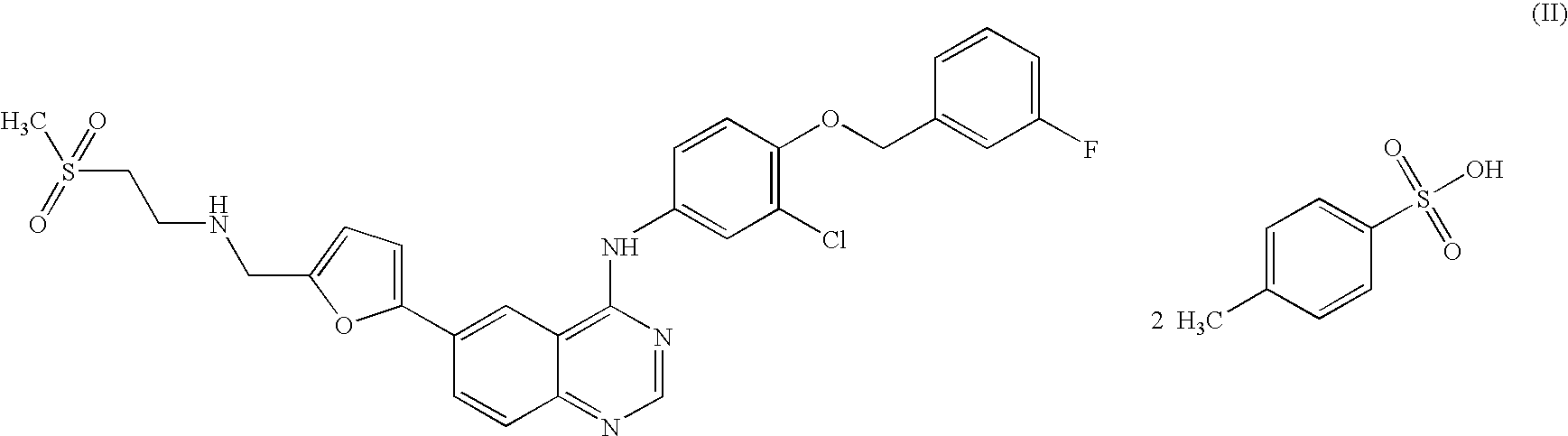
Lapatinib of Formula-(II), was first disclosed by SmithKline Beecham in US Patent No. 6,727,256.
![Figure imgf000002_0001]()
The process for the preparation of Lapatinib of Formula-(II), disclosed in W099/35146, is given in the Scheme-I. Accordingly, 4-chloro-6-iodo-quinazoline of Formula-(IV), is reacted with 3-chloro-4-(3′-fluoro-benzyloxy)-aniline yielding N-[3- chloro-4-{(3'-fluorobenzyloxy) phenyl} ]-6-iodo-quinazoline of Formula-( l). The compound of the Formula-(l) reacts with 5-(l,3-dioxolan-2-yl)-2-(tributylstannyl)furan to get the compound of Formula-(2) which on deprotection with HC1, removes the 1,3- dioxolan-2-yl protecting group and liberates 5-(4-{3-chloro-4-(3-fluoro- benzyloxy)anilino}-6- quinazolinyl)-furan-2-carbaldehyde of Formula-(3). The compound of the Formula-(3) on reaction with 2-methanesulfonylethylamine, followed by reductive amination using sodium (triacetoxy)borohydride as the reducing agent gives the required compound Lapatinib of Formula-(II) as an organic residue, which is purified by column chromatography and subsequently converted into its hydrochloride salt (5).
![Figure imgf000003_0001]()
Subsequently, US 7, 157,466 also discloses the preparation of Lapatinib and its ditosylate salt, which is given in Scheme-II.
Lapatinib ditosylate has been prepared by reacting the tosylate salt of 5-(4-[3- chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde of Formula (3) with 2-(methylsulfonyl)ethylamine in the presence of base (diisopropyl- ethylamine) followed by reduction with sodium triacetoxyborohydride to obtain Lapatinib base which is converted to Lapatinib ditosylate anhydrate by adding para- toulenesulfonic acid. Conversion to Lapatinib ditosylate monohydrate is carried out using THF/H20. Intercon vers ion to the anhydrate of the ditosylate salt and back to monohydrate is carried out with methanol and water respectively.
(lla)
WO201 1039759, filed by Natco Pharma also describes a process for the preparation of Lapatinib from 2-amino benzonitrile, as given in scheme-Ill. Firstly, 2- aminobenzonitrile (6) is reacted with iodine monochloride in acetic acid medium to form compound of Formula (7) which is recrystallized from mixture of hexane and toluene. The compound of Formula (1) is reacted with N,N-dimethylformamide dimethy|acetal in an organic solvent such as toluene or xylene to form novel compound of Formula (8). The compound of Formula (7) is then coupled with compound of Formula (8) in presence of acid catalyst such as trifluoroacetic acid, formic acid or acetic acid to form compound of Formula (3). The compound of Formula (3) is the subjected to Suzuki coupling with 5-formyl-2-furyl boronic acid in ethereal solvent in the presence of catalyst selected from palladium (II) acetate, palladium (II) chloride, and palladium on carbon to form aldehyde compound of Formula (4). The compound of Formula (4) is reacted with 2-methanesulphonyl ethylamine or its salt to produce imine compound of Formula (VI) which is reduced with sodium borohydride to form Lapatinib base (II). The crude Lapatinib base is purified by crystallization from organic solvents. The purified Lapatinib base is converted into Lapatinib ditosylate anhydrous by treating Lapatinib base in organic solvent with /7-toluenesulfonic acid monohydrate which is then recrystallized from aqueous alcohol to produce pharmaceutically acceptable Lapatinib ditosylate monohydrate. The process is depicted in Scheme-Ill.
-IH
![Figure imgf000005_0001]()
Lapatinib (II) WO2010017387, filed by Teva relates to Lapatinib intermediates and process for the preparation of Lapatinib base and Lapatinib ditosylate. The application relates to highly pure intermediate of Formula (2), 3-chloro-4-(3-fluorobenzyloxy)aniline which is prepared by reducing a compound of Formula (1), 3-chloro-4-(3- fluorobenzyloxy)nitrobenzene, with iron and ammonium chloride system in the presence of a C1 -C4 alcohol and water at refluxing temperature. The application also relates to highly pure intermediate of Formula (3), N-[3-chloro-4-(3-fluorobenzyloxy)- phenyl]-6-iodoquinazolin-4-amine, which is prepared in one-pot process from compound of Formula (1 ) by reduction using iron and ammonium chloride system in presence of C1 -C4 alcohol and water. The compound of Formula (3) is reacted with 5- formyl-2-furanboronic acid in the presence of a palladium catalyst and a base in a polar organic solvent to obtain Lapatinib aldehyde base, compound of Formula (4). Optionally, Lapatinib aldehyde base is combined with /? oluenesulfonic acid to obtain Lapatinib aldehyde monotosylate, compound of Formula (5). The invention further provides a process for the preparation of Lapatinib base. Lapatinib aldehyde base or its salt is combined with methylsulfonylethylamine or its hydrochloride salt, acetic acid, an inorganic base in an organic solvent and a reducing agent (sodium triacetoxyborohydride) to form Lapatinib base. Lapatinib base is further purified by using organic solvents. Lapatinib base obtained is further converted to Lapatinib ditosylate. The process is depicted in scheme-IV.
Scheme-IV
Example-5
Preparation of Lapatinib Ditosylate
To a stirred mixture of Sodiumtriacetoxyborohydride (0.21 g) in Tetrahydrofuran (THF)(2.4 ml) was added N-(3-Chloro-4-(3-fluorobenzyloxy)phenyl)-6-(5-((2- (methylsulfonyl)ethylimino)- methyl)furan-2-yl)quinazolin-4-amine (0.2 g) in THF. The reaction mixture was stirred for 1 hour at 20-25 °C. Reaction was monitored by TLC and on completion of reaction, aqueous NaQH (0.16 g NaOH to 0.8 g demineralized water) was added. The organic layer was separated and added p- Toluenesulfonic acid (0.42) in THF (0.6 ml) and stirred for 3 hours. The solid was filtered and dried under vacuum at 60-65°C till constant weight.
Weight: 0.15 g
Yield: 46.9 %
Purity by HPLC: 96.16%
MS (ES+) m/z: 581 [M+H]+ & 583 [M+H+2]+
1H NMR (400 MHz; DMSO-d6): 2.28 (s, 6H), 3.14 (s, 3H), 3.44 (t, J=8.0 Hz, 2H), 3.55 (t, J=8.0 Hz, 2H), 4.46 (s, 2H), 5.31 (s, 2H), 6.89 (br s, 1H), 7.10 (d, J=7.2 Hz, 4H), 7.20 (m, 1H), 7.23 (br s, 1H), 7.31- 7.36 (m, 3H), 7.47 (d, J=7.2 Hz, 4H), 7.63 (d, J=8.8 Hz, IH), 7.89 (br s, IH), 7.92 (d, J=8.8 Hz, IH), 8.39 (d, J=8.8 Hz, IH), 8.89 (s, IH), 8.98 (s, IH), 9.28 (s, IH, NH), 11.18 (s, IH, NH).
………………………………………….
http://www.google.com/patents/WO2008024439A2?cl=en
![]()
…………………………………………..
http://www.google.co.in/patents/US7157466
The free base and HCl salts of the compounds of Formulae (I), (II), (III), and (IV), may be prepared according to the procedures of International Patent Application No. PCT/EP99100048, filed Jan. 8, 1999, and published as WO 99/35146 on Jul. 15, 1999, referred to above. A schematic of such procedures is presented in Scheme A following. The specific page references given are to WO 99/35146. The free base of the compound of formula II is used as an example of the general scheme.
![Figure US07157466-20070102-C00005]()
![Figure US07157466-20070102-C00006]()
The compound of formula (II), i.e., N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl) ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate has been prepared in two distinct forms, an anhydrate form (Formula II′ in Scheme B) and a monohydrate form (Formula II″ in Scheme B). The relationship of these forms is illustrated in Scheme B below. The anhydrate form of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate may be prepared by (a) reacting the tosylate salt of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde (formula B in Scheme B) with 2-(methylsulfone)ethylamine in tetrahydrofuran in the presence of diisopropyl-ethylamine followed by (b) the introduction of this solution into to a slurry of sodium triacetoxyborohydride in tetrahydrofuran at room temperature, (c) adding 5N sodium hydroxide to adjust the pH to within a range of 10–11, (d) separating the organic tetrahydrofuran phase, and then (e) adding para-toulenesulfonic acid hydrate to the organic phase to provide the ditosylate anhydrate. Interconversion to the monohydrate and back to the anhydrate of the ditosylate salt compounds of the invention is as depicted in Scheme B. The tosylate salt of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde is prepared from the HCl salt of the carbaldehyde (Formula A of Scheme B). Preparation of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate and the anhydrate and monohydrate forms thereof are utilized as an example. As recognized by those skilled in the art, other compounds of formula I and anhydrate and hydrate forms thereof may be prepared by similar methods.
![Figure US07157466-20070102-C00007]()
Compound A of Scheme B may be prepared by various synthetic strategies, other that the strategy recited in Scheme A above, utilizing the palladium(O) mediated coupling of quinazoline and substituted furan intermediates.
Example 8
Preparation of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate anhydrate (Anhydrate Form of Compound of Formula II)
To a 20 L reactor was added 13.3 vol of THF followed by 0.62 wt (2.93 mol) of NaBH(OAc)3. The 20 L reactor was set to maintain contents at 20° C. A second 20 L reactor was charged with 1000 grams, (1.55 mol) of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde 4-methyl benzenesulfonate prepared by the procedure of Example 7 and 6.7 vol of THF. To the THF solution of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde 4-methylbenzenesulfonate was added 0.325 vol (1.86 mol) diisopropylethylamine followed by 0.32 wt of 2-(methylsulfone)ethylamine, (321 g, 2.6 mol) and 0.15 vol of IPA. After 1 hour, the preformed imine/THF solution was transferred by vacuum to the stirred suspension of NaBH(OAC)3 in the first 20 L reactor over 10 minutes. After 90 minutes, 4 vol of 5N NaOH was added over 40 min via a pump. This solution was allowed to stir for 15 minutes after which the stirrer was switched off and the layers were allowed to separate. The aqueous layer was drained from the bottom of the reactor and the organic layer transferred to the empty 20 L reactor through a teflon-lined stainless steel jacketed transfer hose outfitted with an in-line 0.45 μm filter. To this solution was added a 2 vol THF solution of 4 wt (1180 g, 6.2 mole) of p-toluenesulfonic acid monohydrate over 5 min. A yellowish precipitate was observed to come out of solution and this was allowed to stir at room temperature for 12 hours. The reaction was drained from the bottom of the reactor and filtered through a ceramic filter lined with paper. The yellow filter cake was washed with 1 vol of a 95:5 THF water solution and allowed to air dry overnight. After suctioning dry for 12 hours, the yellow filter cake was transferred to two glass trays and placed in the drying oven (42° C.) under house vacuum (18 in Hg) with a nitrogen bleed. The two glass trays were removed from the oven and allowed to cool to room temperature and sampled accordingly. The isolated yield of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methane-sulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate (anhydrate) was 1264 grams (1.3 wt, 88%; 1443 g Th) and was a yellow solid.
Approximately 50 mg of the product was transferred to a Karl Fisher Volumetric Moisture Apparatus (model DL35, Mettler, Hightstown, N.J.), which was operated according to the manufacturer’s instructions. The anhydrate water content was determined to be 0.31%.
Example 10Preparation of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate monohydrate (Monohydrate Form of Compound of Formula II)
A 20 L reactor was charged with 1 wt (930 g, 1.0 mol) of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate anhydrate prepared using the procedure of Example 8. To this was added 10 volumes of a pre-mixed 8:2 THF:deionized water solution and the reactor was heated to 65° C. Complete dissolution was observed at 50° C. The clear reaction mixture was transferred to another 20 L reactor through a stainless steel jacketed transfer hose that was equipped with an in-line 5.0 μm cartridge filter. The empty 20 L reactor and the filter line were washed with 0.2 vol of the pre-mixed 8:2 THF:deionized water solution. An additional 1 vol of pre-mixed 8:2 THF:deionized water solution was used to wash the material into the reaction mixture. The 20 L reactor was heated to ˜80° C. The reaction temperature was then ramped down to 55° C. over 2 hours and then to 45° C. over 10 hours. After 10 hours, the temperature was adjusted to 25° C. and the reaction mixture allowed to stir at room temperature for 45 minutes. The yellow precipitate was drained from the bottom of the 20 L reactor into a ceramic filter lined with paper. The flow was fast and smooth and the filter rate very good. The yellow filter cake was washed with 0.6 volumes of a pre-mixed 8:2 THF:deionized water solution and the yellow solid was air dried for 4 hours and placed into a glass tray. The glass tray was placed in a vacuum oven under house vacuum (˜18 in Hg) at 60° C. with a nitrogen bleed for 2 days. After removal from the oven, the material was sampled accordingly. The yield was 743 grams (0.8 wt, 80%; 930 g th) as a bright yellow, crystalline solid.
Approximately 50 mg of the product was transferred to a Karl Fisher Volumetric Moisture Apparatus (model DL35, Mettler, Hightstown, N.J.), which was operated according to the manufacturer’s instructions. The monohydrate water content was determined to be 1.99%, which is in agreement with the theoretical value of 1.92%.
![Figure US07157466-20070102-C00002]()
Literature References:
Reversible dual inhibitor of ErbB1 and ErbB2 tyrosine kinases. Prepn: M. C. Carter et al., WO 9935146(1999 to Glaxo); eidem, US6727256 (2004 to SmithKline Beecham).
Mechanism of action study: W. Xia et al., Oncogene 21, 6255 (2002); and crystal structure in complex with epidermal growth factor receptor (EGFR, ErbB1): E. R. Wood et al., Cancer Res. 64, 6652 (2004).
In vitro antitumor activity in combination with anti-ErbB2 antibodies: W. Xia et al., Oncogene 24, 6213 (2005). Biologic effects on tumor growth: N. L. Spector et al., J. Clin. Oncol. 23, 2502 (2005).
Pharmacokinetics and clinical activity in metastatic carcinomas: H. A. Burris III et al., ibid. 5305.
Review of clinical development: T. E. Kim, J. R. Murren, IDrugs6, 886-893 (2003); H. A. Burris III, Oncologist 9, Suppl. 3, 10-15 (2004).
Lapatinib Ditosylate [USAN]
![]()
- Lapatinib ditosylate monohydrate
- Tykerb
- Tyverb
- UNII-G873GX646R
- KS-1300; 388082-78-8
![Chemical structure for LAPATINIB DITOSYLATE MONOHYDRATE]()
Dosages/Routes/Forms
Dosages/Routes/Forms
| Drug Name |
Active Ingredient(s) |
Strength |
Form/Route |
Marketing Status |
RLD |
TE Code |
| TYKERB |
LAPATINIB DITOSYLATE |
EQ 250MG BASE |
TABLET;ORAL |
1 |
1 |
|
Approval History
2013-10-18
017
Efficacy Supplement with Clinical Data to Support
2010-01-29
007
New or Modified Indication New or Modified Indication
Derivative Type: Ditoluenesulfonate monohydrate
CAS Registry Number: 388082-78-8; 388082-77-7 (anhydrous)
Additional Names: Lapatinib ditosylate
Manufacturers’ Codes: GW-572016F
Molecular Formula: C29H26ClFN4O4S.2C7H8O3S.H2O
Molecular Weight: 943.48
Percent Composition: C 54.74%, H 4.70%, Cl 3.76%, F 2.01%, N 5.94%, O 18.65%, S 10.20%
Properties: Yellow solid.
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Tyrosine Kinase Inhibitors.
References
- Burris HA (2004). “Dual kinase inhibition in the treatment of breast cancer: initial experience with the EGFR/ErbB-2 inhibitor lapatinib”. Oncologist. 9 Suppl 3: 10–5.doi:10.1634/theoncologist.9-suppl_3-10. PMID 15163842.
- Higa GM & Abraham J (September 2007). “Lapatinib in the treatment of breast cancer”. Expert Review of Anticancer Therapy (log in required) (Future Drugs) 7(9): 1183–92. doi:10.1586/14737140.7.9.1183. PMID 17892419.
- Pazdur, Richard (14 January 2011). “FDA Approval for Lapatinib Ditosylate”.Womens Health (Lond Engl) (Cancer.gov) 6 (2): 173. doi:10.2217/whe.10.11.PMID 20187722.
- ^ Jump up to:a b c d “GlaxoSmithKline receives marketing authorisation in the EU for Tyverb (lapatinib), the first oral targeted therapy for ErbB2-positive breast cancer” (Press release). GlaxoSmithKline. 2008-06-12. Retrieved 2008-06-21.
- ^ Jump up to:a b c “GlaxoSmithKline Reports Positive New Data On Tykerb (lapatinib) At The 2007 American Society Of Clinical Oncology (ASCO) Annual Meeting” (Press release). Medical News Today. June 4, 2007. Retrieved December 2, 2008.
- Jump up^ “Data Sheet: TYKERB”. Medsafe. New Zealand Medicines and Medical Devices Safety Authority. March 12, 2008. Retrieved December 2, 2008.
- Jump up^ Kulkarni, Kaustubh (2 August 2013). “India revokes GSK cancer drug patent in latest Big Pharma blow”. Reuters (Mumbai, India: Reuters). Retrieved 2 August 2013.
- Jump up^ Wood, ER, Truesdale, AT, McDonald, OB, Yuan, D, Hassell, A, Dickerson, SH, Ellis, B, Pennisi, C et al. (2004). “A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells”. Cancer Research 64 (18): 6652–9. doi:10.1158/0008-5472.CAN-04-1168. PMID 15374980.
- Jump up^ Dr. Angel Rodriguez (April 2008). “New type of drug shrinks primary breast cancer tumors significantly in just six weeks; research provides leads to a new target in cancer treatment – the cancer stem cell”.
- Jump up^ Nelson MH, Dolder CR (February 2006). “Lapatinib: a novel dual tyrosine kinase inhibitor with activity in solid tumors”. Ann Pharmacother 40 (2): 261–9.doi:10.1345/aph.1G387. PMID 16418322.
- Jump up^ Geyer CE, Forster J, Lindquist D, et al. (December 2006). “Lapatinib plus capecitabine for HER2-positive advanced breast cancer”. N. Engl. J. Med. 355 (26): 2733–43.doi:10.1056/NEJMoa064320. PMID 17192538.
- J Burris HA, Hurwitz HI, Dees EC, et al. (August 2005). “Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas”. J. Clin. Oncol. 23 (23): 5305–13.doi:10.1200/JCO.2005.16.584. PMID 15955900.
- J NCI Cancer Drug Information. FDA Approval for Lapatinib Ditosylate (Tykerb®). Retrieved 27 January 2014.
- |url=http://www.bioportfolio.com/news/article/1492867/GSK-Tykerb-Tyverb-Phase-III-gastric-cancer-study-fails-to-meet-primary.html
External links
| WO1999035146A1 |
Jan 8, 1999 |
Jul 15, 1999 |
Glaxo Group Ltd |
Bicyclic heteroaromatic compounds as protein tyrosine kinase inhibitors |
| WO2010017387A2 |
Aug 6, 2009 |
Feb 11, 2010 |
Teva Pharmaceutical Industries Ltd. |
Lapatinib intermediates |
| WO2011039759A1 |
Sep 29, 2009 |
Apr 7, 2011 |
Natco Pharma Limited |
A new process for the preparation of lapatinib and its pharmaceutically acceptable salts |
| US6727256 |
Jan 8, 1999 |
Apr 27, 2004 |
Smithkline Beecham Corporation |
4-aminoquinazoline derivatives as anticarcinogenic agents |
| US7157466 |
Jun 28, 2001 |
Jan 2, 2007 |
Smithkline Beecham (Cork) Limited |
Quinazoline ditosylate salt compounds |
| WO1998002434A1 * |
Jul 11, 1997 |
Jan 22, 1998 |
Malcolm Clive Carter |
Fused heterocyclic compounds as protein tyrosine kinase inhibitors |
| WO2007121279A2 * |
Apr 12, 2007 |
Oct 25, 2007 |
Tona Morgan Gilmer |
Cancer treatment method |
Filed under:
cancer Tagged:
lapatinib ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()


 just an animation
just an animation



























 Originally posted on
Originally posted on 
























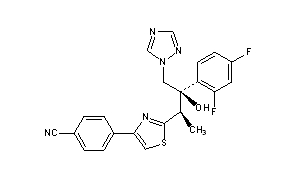

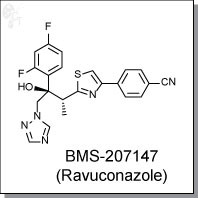




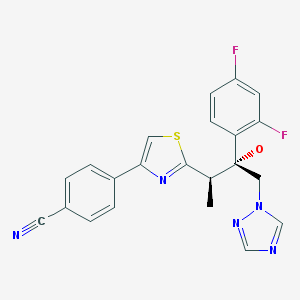
























































































































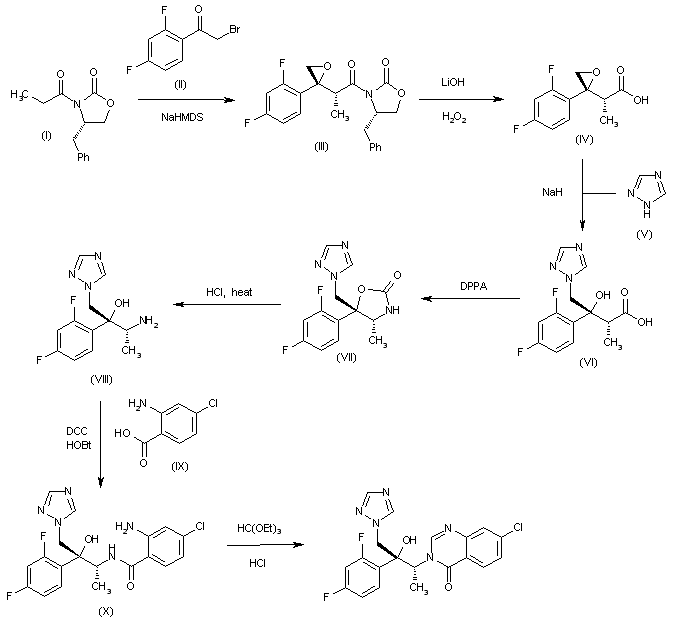

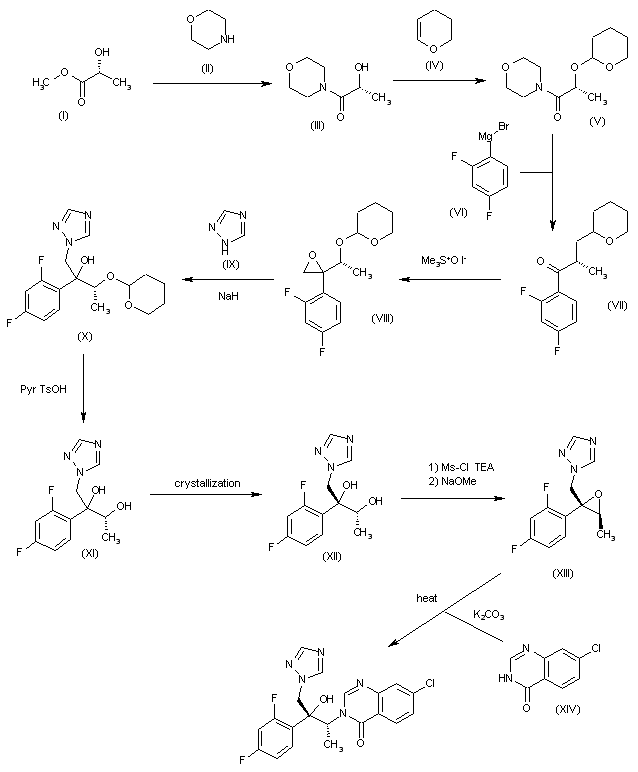

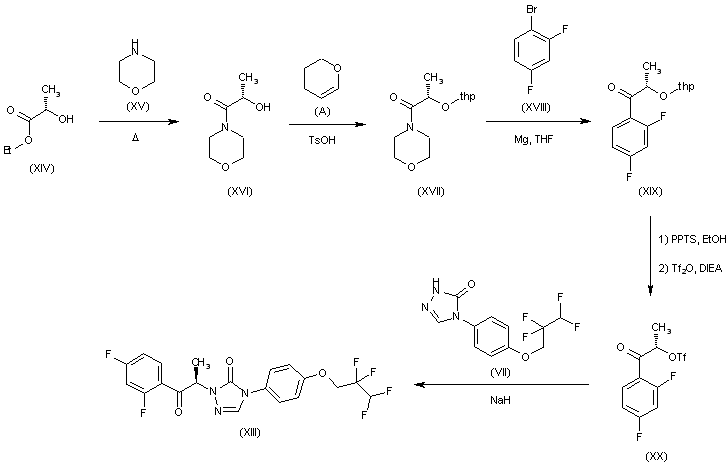

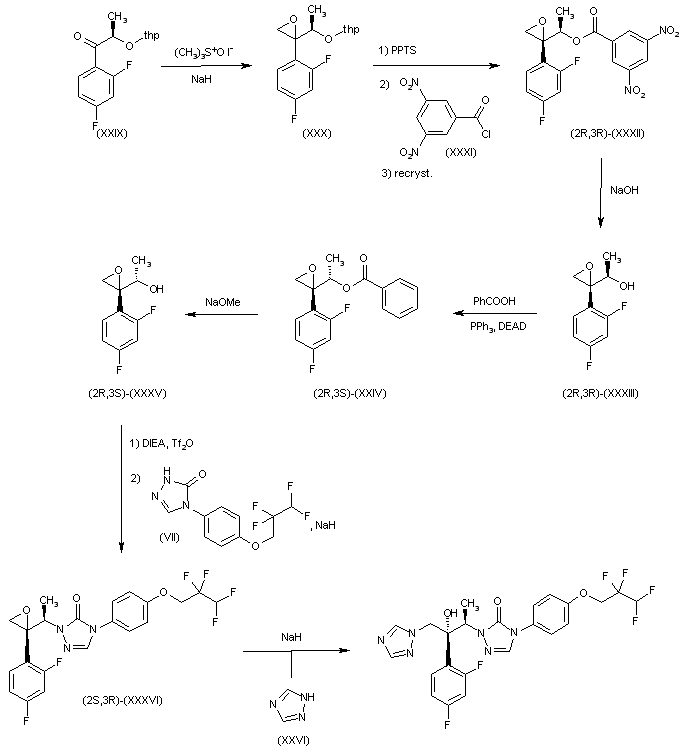
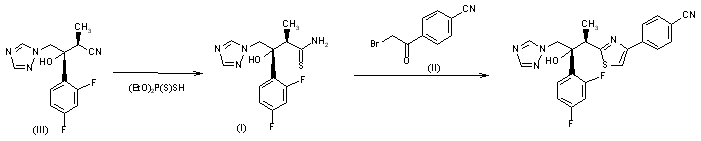
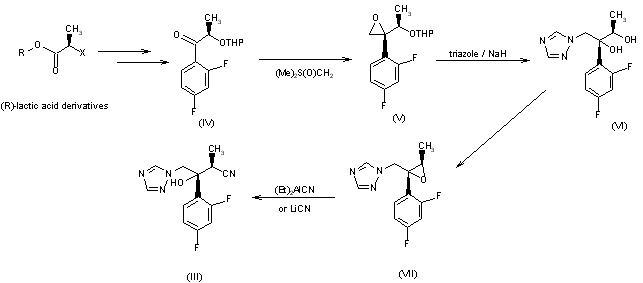
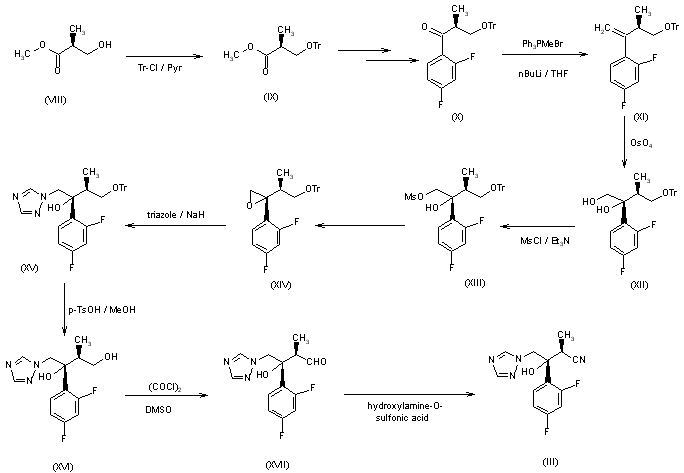
 CH-N), 8.26 (d, J = 8.6, 1H, arom), 8.11 (d, J = 5.7, trace rotamer), 7.76 (s, 2H, triazol),
CH-N), 8.26 (d, J = 8.6, 1H, arom), 8.11 (d, J = 5.7, trace rotamer), 7.76 (s, 2H, triazol), 























































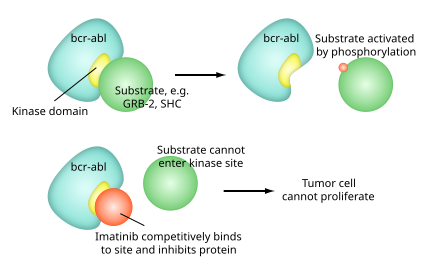
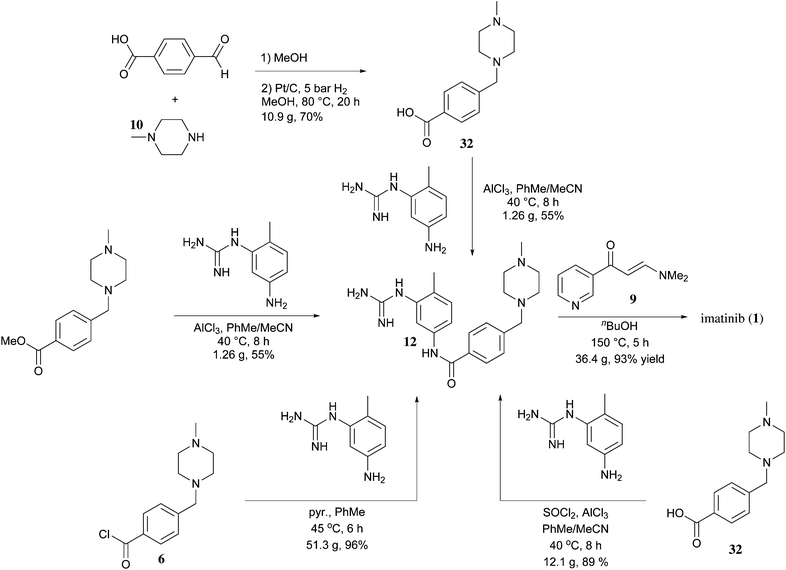
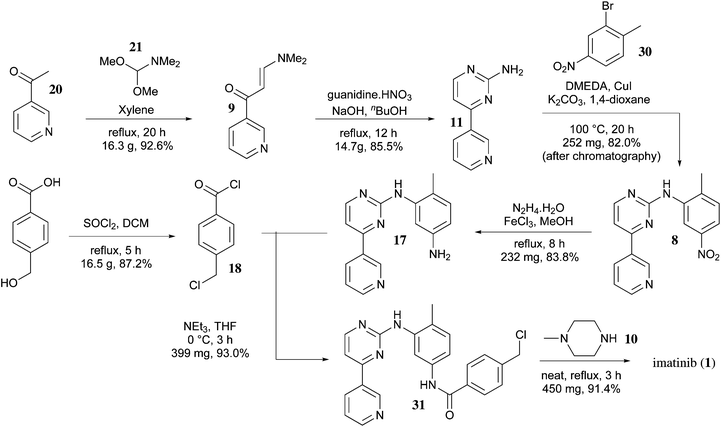
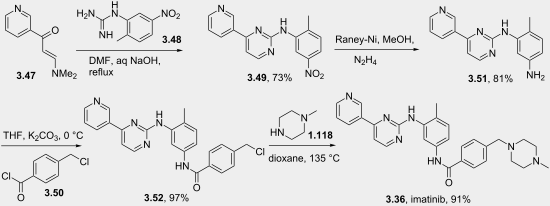
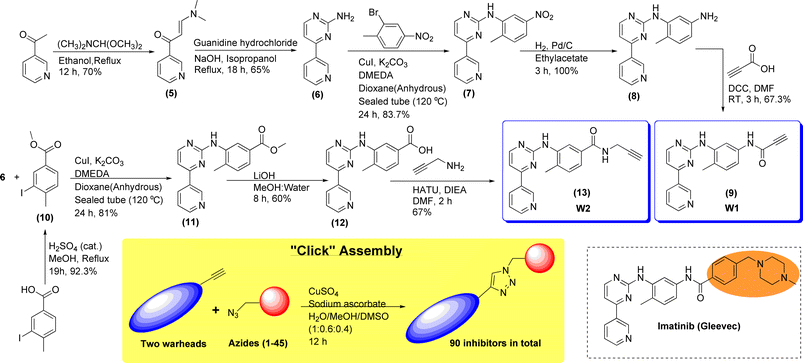
 Imatinib
Imatinib
























![[1860-5397-9-265-i38]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i38.png?max-width=550&background=EEEEEE)



































































![[1860-5397-9-265-i41]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i41.png?max-width=550&background=EEEEEE)